46
Содержание:
- Накопление вредных мутаций на Y-хромосоме
- Шаблон наследования
- Хромосомы у человека. Хромосомные болезни
- Аномалии генетического материала
- Синдром Шерешевского-Тернера
- Что такое микроделеционный синдром?
- Симптомы андрогенной недостаточности при синдроме Клайнфельтера
- Половой хроматин в судебно-медицинском отношении
- Реабилитация юной Y-хромосомы
- СИНДРОМ КЛЯЙНФЕЛЬТЕРА
- Синдром Ди Джорджи
- Регуляция пола
- Аниридия
- Половые хромосомы у цветковых растений
- Количество в различных организмах
Накопление вредных мутаций на Y-хромосоме
Считается, что эволюция половых хромосом происходит через мутация аутосом, которые несут гены определения пола. В какой-то момент, когда происходит кластеризация генов для определения пола на одной из двух аутосом, происходит подавление рекомбинации, чтобы гарантировать, что кластер генов наследуется в одном блоке. Однако, как только это происходит, зарождающаяся Y-хромосома начинает формироваться, накапливая переносимые элементы, хромосомные перестройки и другие вредные мутации, которые путешествуют автостопом с благоприятными генами определения пола. Сказано, что это приводит к полностью гетероморфным половым хромосомам и определению пола.
Неспособность Y-хромосомы автоматически исправлять мутации путем рекомбинации во время мейоза делает ее особенно склонной к накоплению ошибок. Кроме того, сперматозоиды образуются в большом количестве, вовлекая многие деление клеток события, которые увеличивают вероятность накопления ошибок. Сперма также сохраняется в сильно окислительной среде в яичках, что снова увеличивает вероятность генетической мутации. Одна из гипотез гласит, что эти факторы способствовали возникновению ситуации, когда Y-хромосома потеряла большинство своих генов, за исключением тех, которые имеют решающее значение для определения пола и выживания плод, Это приводит к тому, что у гомогаметных женщин число генов в их половых хромосомах почти удваивается по сравнению с их гетерогаметными партнерами. У некоторых животных, у которых самцы имеют XY, экспрессия генов на одной из Х-хромосом у самок заглушается посредством образования гетерохроматина, Альтернативно, некоторые насекомые предпочитают сверхэкспрессировать гены на своей Х-хромосоме у гетерогаметных особей. Эта модификация экспрессии генов называется дозовой компенсацией. В последнее время впервые была отмечена компенсация дозировки в двудомном растении S.latifolia или во время кампионии.
Шаблон наследования
Число возможных предков по линии наследования Х-хромосомы в данном предковом поколении соответствует последовательности Фибоначчи. (После Хатчисона, Л. «Выращивание семейного древа: сила ДНК в восстановлении семейных отношений».)
Люк Хатчисон заметил, что ряд возможных предков по линии наследования Х-хромосомы в данном предковом поколении следует последовательности Фибоначчи . У мужчины есть Х-хромосома, которую он получил от своей матери, и Y-хромосома , которую он получил от своего отца. Самец считается «источником» его собственной Х-хромосомы ( ), а в поколении его родителей его Х-хромосома произошла от одного родителя ( ). Мать мужчины получила одну Х-хромосому от своей матери (бабушка по материнской линии сына) и одну от своего отца (деда по материнской линии), поэтому двое бабушек и дедушек внесли свой вклад в Х-хромосому потомка мужского пола ( ). Дед по материнской линии получил свою Х-хромосому от своей матери, а бабушка по материнской линии получила Х-хромосомы от обоих своих родителей, поэтому три прабабушки и дедушки внесли свой вклад в Х-хромосому мужского потомка ( ). Пять прапрапрадедов внесли свой вклад в Х-хромосому мужского потомка ( ) и т. Д
(Обратите внимание, что это предполагает, что все предки данного потомка независимы, но если какая-либо генеалогия прослеживается достаточно далеко во времени, предки начинают появляться на несколько строк генеалогии, пока, в конце концов, основатель населения не появится во всех строках генеалогии.)
F 1 знак равно 1 {\ displaystyle F_ {1} = 1} F 2 знак равно 1 {\ displaystyle F_ {2} = 1} F 3 знак равно 2 {\ displaystyle F_ {3} = 2} F 4 знак равно 3 {\ displaystyle F_ {4} = 3} F 5 знак равно 5 {\ displaystyle F_ {5} = 5}
Хромосомы у человека. Хромосомные болезни
Хромосомные болезни — наследственные заболевания, обусловленные изменением числа или структуры хромосом.
К хромосомным относятся болезни , обусловленные геномными мутациями или структурными изменениями отдельных хромосом. Хромосомные болезни возникают в результате мутаций в половых клетках одного из родителей. Из поколения в поколение передаются не более 3—5 % из них. Хромосомными нарушениями обусловлены примерно 50 % спонтанных абортов и 7 % всех мёртворождений.
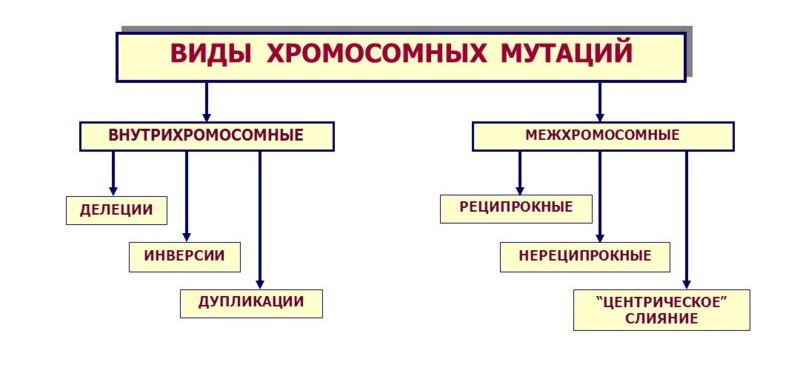
Все хромосомные болезни принято делить на две группы: аномалии числа хромосом и нарушения структуры хромосом.
Основная статья: Хромосомные перестройки
В настоящее время у человека известно более 700 заболеваний, вызванных изменением числа или структуры хромосом. Около 25 % приходится на аутосомные трисомии, 46 % — на патологию половых хромосом. Структурные перестройки составляют 10,4 %. Среди хромосомных перестроек наиболее часто встречаются транслокации и делеции.
Аномалии генетического материала
Наследственный материал состоит из огромного количества нуклеотидов, формирующих гены. При этом в каждом гене последовательность нуклеотидов строго определена, поскольку должна кодировать определенный белок. Кроме того, сами гены при формировании хромосом также выстраиваются в фиксированном порядке. Благодаря сохранению этого порядка организм может функционировать, а ученые – быстро и точно указывать друг другу, про какой ген идет речь.
В идеальном случае система работает без малейших сбоев, а генетическая информация всегда передается в неизменном виде. Однако на практике большое число структурных единиц и постоянное воздействие различных факторов (например, ионизирующего излучения) приводит к тому, что время от времени возникают различные аномалии. В частности, отдельные участки последовательности ДНК могут быть скопированы на новое место. В таком случае говорят о дупликации. Если же вместо создания новой копии была перемещена часть исходной цепочки, то модификация называется транслокацией. Кроме того, иногда часть последовательности просто теряется, удаляется из генетического материала. В таком случае изменение называется делецией.
Поскольку взаимодействия в организме оттачивались в течение многих тысячелетий эволюционного развития, получилась очень слаженная система. И аномалии, даже самые небольшие, могут вызвать нарушение баланса. В таком случае в организме развивается то или иное нарушение. Если при этом причина находится на уровне генов, то говорят о генных болезнях. Если была утрачена или наоборот получена лишняя копия хромосомы, то такие нарушения называются хромосомными заболеваниями.
Синдром Шерешевского-Тернера
Является следствием моносомии по хромосоме Х. Больные дети часто рождаются недоношенными или имеют сниженную массу тела. Одним из классических признаков синдрома Шерешевского-Тернера, который можно заметить сразу после рождения, является выраженная кожная складка на шее. Среди других клинических проявлений отмечаются:
- пороки сердца;
- отечность верхних и нижних конечностей;
- нарушение циркуляции лимфы;
- задержка речевого и физического развития.
По мере взросления ребенка проявляются характерные черты строения тела. Рост обычно не превышает 150 см, крыловидные складки на шее сохраняются, ушные раковины могут деформироваться, верхняя челюсть недоразвита, грудная клетка широкая. Моносомия по хромосоме Х влияет на развитие органов половой системы. У женщин отмечается отсутствие фолликулов в яичниках, нарушение менструального цикла, недоразвитие молочных желез. У мужчин снижается уровень тестостерона, может отсутствовать одно или оба яичка либо отмечаться их недоразвитие.
Прогноз при синдроме Шерешевского-Тернера относительно благоприятный. При отсутствии тяжелых пороков развития и регулярном наблюдении у специалиста продолжительность жизни не сокращается.
Что такое микроделеционный синдром?
Самые незначительные изменения (они же мутации) называются точечными. Их появление влияет на считанные единицы генов. В некоторых случаях нарушение относится вообще к одному единственному гену
Однако если он обеспечивал выработку важного белка, последствия для всего организма могут быть очень серьезными. Подобные патологические изменения относятся к группе микроделеционных синдромов
Каждое такое заболевание обусловлено небольшим изменением генетического материала, которое происходит в строго определенном месте. Точный механизм возникновения подобных нарушений на сегодняшний день не установлен, что не мешает ученым заниматься исследованием их воздействия на организм.
Так, было выяснено, что развитие синдрома в таком случае может происходить несколькими различными вариантами. В частности, ряд заболеваний характеризуется участием онкогенов. В других случаях на воздействие непосредственно самой делеции накладывается эффект хромосомного импринтинга и возможные однородительские дисомии.
Частота возникновения большей части микроделеционных синдромов крайне невелика: порядка 1 случая на 50-100 тысяч новорожденных. Набор клинических признаков обычно выражается отчетливо. Для того чтобы поставить диагноз, бывает достаточно лишь совокупности симптомов. Однако при таком подходе невозможно точно прогнозировать здоровье потомков, поэтому зачастую наряду с проверкой обычных признаков производится молекулярно-генетическая диагностика пробанда и его родственников (обычно родители, в некоторых случаях также требуется анализ генотипа братьев, сестер, теть, дядь и так далее).
Патологические проявления сильно отличаются. В частности, их проявление определяется тем, насколько большой участок генетического материала был утрачен в результате делеции. Кроме того, в ряде случаев играет роль то, от кого из родителей была получена мутация (влияние хромосомного импринтинга). Хорошей иллюстрацией последней ситуации является пара синдромов Прадера-Вилли и Ангельмана. Они оба обусловлены наличием делеции в 15 хромосоме. Однако из-за различного механизма действия при передаче от разных родителей клиническая картина этих заболеваний значительно отличается.
Симптомы андрогенной недостаточности при синдроме Клайнфельтера
Андрогенная недостаточность при синдроме Клайнфельтера связана с постепенной атрофией яичек, что приводит к снижению синтеза тестостерона. Степень недостаточности андрогенов резко варьирует.
В первую очередь обращают на себя внимание внешние признаки гипогонадизма:
- скудная растительность на лице или же полное ее отсутствие;
- рост волос на лобке по женскому типу;
- волосы на груди и других частях тела отсутствуют;
- маленький объем яичек (2–4 мл) и их плотная консистенция (патогномоничный признак).
Поскольку дегенерация половых желез, как правило, развивается в постпубертатный период, у большинства пациентов размеры мужских половых органов, за исключением яичек, соответствуют возрастным нормам.
Пациенты могут жаловаться на ослабление либидо и снижение потенции. У многих мужчин с синдромом Клайнфельтера половое влечение вовсе не возникает, а некоторые — напротив, заводят семью и живут нормальной половой жизнью. Наиболее постоянный признак патологии — бесплодие, именно оно чаще всего становится причиной обращения таких пациентов к врачу. У 10 % мужчин с азооспемией обнаруживают синдром Клайнфельтера.
Всем пациентам с нарушениями сперматогенеза необходимо определять кариотип для исключения или подтверждения диагноза синдрома Клайнфельтера.
Недостаток андрогенов приводит к развитию остеопороза, анемии и слабости скелетной мускулатуры. У трети больных можно наблюдать варикозное расширение вен голеней.
Андрогены влияют на обмен веществ, поэтому больные с синдромом Клайнфельтера склонны к ожирению, нарушению толерантности к глюкозе и сахарному диабету второго типа.
Доказана предрасположенность таких пациентов к аутоиммунным заболеваниям (ревматоидный артрит, системная красная волчанка, аутоиммунные заболевания щитовидной железы и другие).
Половой хроматин в судебно-медицинском отношении
Исследование П. х. в суд.-мед. практике производится с целью установления половой принадлежности следов крови, слюны и других биол, жидкостей, вырванных волос, следов-отпечатков клеток тканей и органов, кусочков тканей, которые могут быть обнаружены на месте происшествия, на различных предметах, одежде, теле потерпевшего и подозреваемого в совершении преступления, на орудиях травмы, на транспортных средствах, а также при обнаружении обгоревших трупов или частей расчлененных трупов. Реже П. х. исследуют с целью суд.-мед. установления генетического пола у лиц с аномалиями полового развития, используя общепринятые методики.
Для приготовления препаратов из следов крови (см.) и слюны (см.) кусочки предмета-носителя помещают в пробирку и заливают 0,5—40% (следы крови) или 5—10% (следы слюны) уксусной к-той. Экстрагируют при комнатной температуре в течение нескольких часов и, удалив кусочки предмета-носителя, центрифугируют. Осадок переносят на предметное стекло и высушивают на воздухе. С пятен крови на предметах, не впитывающих жидкость (металл, стекло, пластмасса и др.), делают соскобы, которые затем обрабатывают таким же образом.
При исследовании волос (см.) корень волоса помещают на предметное стекло и добавляют 10—25% уксусную к-ту. После набухания отделяют и измельчают волосяной фолликул, удаляя остальные части волоса.
Из кусочков тканей и органов, при необходимости предварительно выдержав их до набухания в уксусной к-те соответствующей концентрации или в физиол, р-ре, готовят гистологические препараты, мазки или препараты-отпечатки. Следы-наложения клеток, тканей или органов на орудиях травмы смывают физиол, р-ром, одновременно соскабливая их. Мелкие кусочки тканей, встречающиеся в таких следах, измельчают препаровальными иглами. Смывы-соскобы помещают в пробирки, центрифугируют, из осадка готовят гистологические препараты. Исследование препаратов целесообразно начинать с выявления Y-хроматина, т. к. при его отсутствии те же препараты могут быть снова использованы для выявления X-хроматина. При исследовании учитывают только достаточно хорошо сохранившиеся неповрежденные ядра клеток. При анализе следов крови Y-хроматин определяют в ядрах лимфоцитов, т. к. в нейтрофилах Y-хроматин в препаратах из следов крови мужчин может не выявляться.
При отсутствии повышенной влажности Половой хроматин может длительно сохраняться в высохших следах, а также в клетках фолликула вырванного волоса. Высокая температура (выше 150°) разрушает ядра клеток и П. х. Значительная влажность в течение нескольких суток также приводит к разрушению клеток, что делает невозможным выявление полового хроматина. Т. к. условия, в к-рых находятся следы, могут последовательно меняться, решающее значение для установления пригодности следов крови, слюны и т. д. для определения П. х. имеет состояние обнаруживаемых в них клеток и их ядер. В клетках высохших кусочков тканей, не подвергающихся действию влаги, П. х. сохраняется длительное время. В целых трупах и в их крупных частях в процессе аутолиза и гниения в течение нескольких суток происходит деструкция клеточных ядер. В обгоревших трупах половой хроматин нек-рое время может сохраняться в клетках глубоко расположенных органов и тканей.
При выявлении небольшого числа клеток, сохранивших ядра, исследуемых на Половой хроматин, с целью установления статистической достоверности результатов используют различные математические методы анализа, учитывающие как общее число обнаруженных клеток, так и число клеток. содержащих X- или Y-хроматин.
Библиография: Давиденкова Е. Ф., Берлинская Д. К. и Тысячнюк С. Ф. Клинические синдромы при аномалиях половых хромосом, Л., 1973; Захаров А. Ф. Хромосомы человека, М., 1977; Капустин А. В. Судебно-медицинская диагностика пола по половым различиям в клетках, М., 1969; Лабораторные и специальные методы исследования в судебной медицине, под ред. В. И. Пашковой и В. В. Томилина, с. 157, М., 1975; Любинская С. И. и Антонова С. Н. Исследование Y-хроматина в следах крови, Суд.-мед. экспертиза, т. 18, № 3, с. 17, 1975; Основы цитогенетики человека, под ред. A. А. Прокофьевой-Бельговской, М., 1969; Methods in human cytogenetics, ed. by H. G. Schwarzacher a. U. Wolf, p. 207, B. а. о., 1974; The sex chromatin, ed. by K. L. Moore, Philadelphia — L., 1966.
Реабилитация юной Y-хромосомы
Чтобы устранить эти неувязки, мы решили изучить половые хромосомы гуппи с помощью иммуноокрашивания ключевых белков мейоза (такой метод раньше не применялся в исследованиях этого вида). Из семенников гуппи приготовили препараты мейотических клеток и нанесли на них меченные флуоресцентными красителями антитела к двум белкам — SYCP3 и MLH1. Первый белок образует оси хромосом, а второй маркирует точки рекомбинации. ДНК мы окрасили синим флуоресцентным красителем DAPI. В результате нам удалось получить изображение бивалентов (спаренных хромосом) с отмеченными на них точками рекомбинации (рис. 5, а). В каждой клетке было 23 бивалента, и каждый имел по крайне мере одну точку рекомбинации. Какой же из них образован X- и Y-хромосомами и где у XY-бивалента проксимальный конец, а где дистальный, т. е. где находятся центромеры составляющих его хромосом?
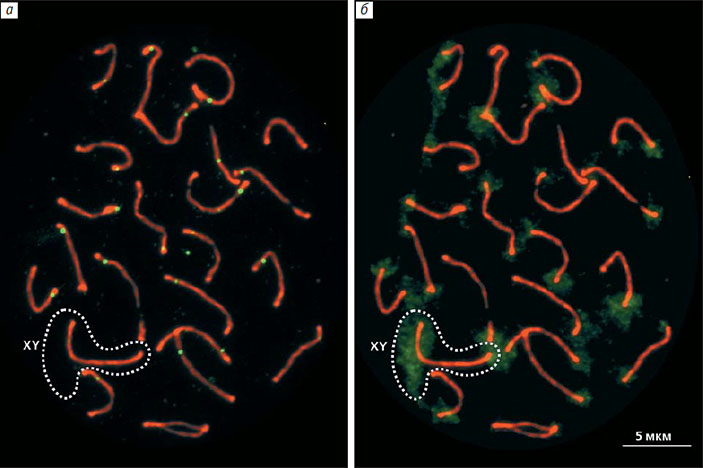
Для идентификации XY-бивалента использовали флуоресцентную in situ гибридизацию ДНК самца гуппи с его мейотическими хромосомами. Мы нанесли на препараты хромосом помеченную зеленым флуорохромом пробу ДНК и с помощью этого метода выявили гетерохроматиновые (содержащие сильно уплотненную ДНК) районы хромосом. Это были блоки прицентромерного гетерохроматина на всех хромосомах, включая и X, и Y, а также очень большой гетерохроматиновый блок на дистальном конце полового бивалента (рис. 6, б). В половых бивалентах с незавершенным синапсисом меченый дистальный сегмент был спарен, а проксимальные концы X и Y хромосом оставались свободными (рис. 6, а, б). Именно так выглядело большинство половых бивалентов с незавершенным синапсисом.
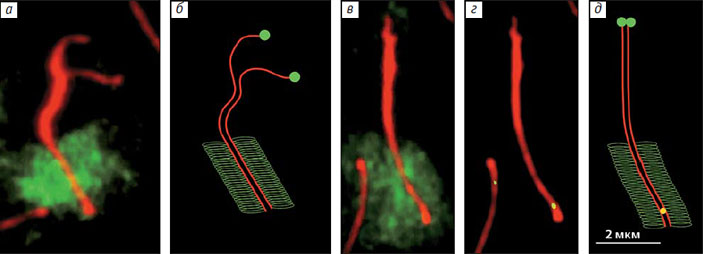
Таким образом, мы установили, что у гуппи спаривание половых хромосом начинается с дистального конца, а не с проксимального, как полагали ранее
Так ли это важно, с какого конца начинается синапсис, если по его завершении хромосомы все равно оказываются спаренными по всей длине и, казалась бы, рекомбинация возможна? В том-то и дело, что, согласно современным представлениям, это очень важно, ведь рекомбинация только и может происходить в точках инициации синапсиса, поскольку она его и инициирует (см. рис. 1, зиготена). Точки рекомбинации мы наблюдали почти на всех половых бивалентах, а не только на 5% из них, как предсказывали генетические данные
Точки эти показывали очень интересное распределение: в проксимальной половине XY-бивалента их вовсе не было, совсем мало (около 5%) — примерно в его середине, а все остальные концентрировались в дистальной четверти бивалента (рис. 6, в, г). Между этими двумя районами находилась зона, запретная для рекомбинации. Преимущественная локализация обменов на конце вполне согласуется с давно обнаруженным соединением концов половых хромосом на поздних стадиях мейоза. Этот факт подтвердили и наши исследования.
Теперь концы с концами вроде бы сходятся (рис. 7). Согласно нашей модели, синапсис начинается в зиготене преимущественно с дистальных концов половых хромосом, где в пахитене наблюдается абсолютное большинство точек рекомбинации, а в метафазе I обнаруживаются хиазмы, что гарантирует правильное расхождение хромосом в гаметы. Остается один вопрос — почему проведенный ранее генетический анализ выявил так мало точек рекомбинации между X и Y только в середине бивалента, а на его конце даже не обнаружил их множество? Все дело в том, что как подтвердили последние исследования с использованием FISH, в руках генетиков пока нет маркеров, локализованных на дистальном конце, и исследователи просто не видят происходящих там обменов.
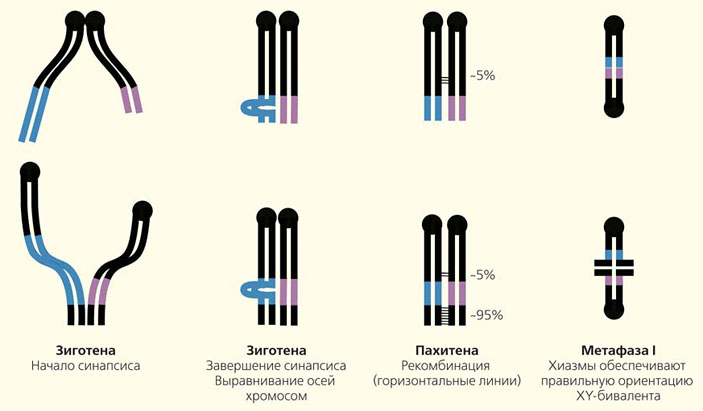
Итак, содержащиеся в Y-хромосоме гуппи ген-детерминатор пола, гены мужских достоинств и примкнувший к ней самцовый блок гетерохроматина скорее всего находятся в запретной для рекомбинации зоне, расположенной в дистальном районе хромосомы. Поскольку зона эта пока невелика, то значит и Y-хромосома изученных нами рыбок еще очень молода и выглядит весьма неплохо.
СИНДРОМ КЛЯЙНФЕЛЬТЕРА
- Кариотип 47,ХХY, тест на половой хроматин – положительный. Y-хроматин –положительный.
- Частота встречаемости 1:500 – 1:700 новорожденных мальчиков.
Фенотип мужской, возможна гинекомастия, евнухоидный тип телосложения (ширина таза обычно больше ширины плеч, удлиненные нижние конечности, высокий рост, скудное оволосение), обычно микроорхидизм при нормальном размере полового члена. Возможен крипторхизм. В редких случаях (при увеличении количества Х-хромосом: 48,ХХХY и 49,ХХХХY) – нарушения интеллекта, функции внутренних органов.
В 5-10% случаев наблюдается мозаичная форма (46,ХY/47,ХХY или 46,ХХ/47,ХХY). В редких случаях у таких больных возможно обнаружить сперматогенез (если клетки сперматогенеза относятся к клону с нормальным кариотипом).
Практически все взрослые больные с синдромом Кляйнфельтера бесплодны вследствие азооспермии.
Учитывая, что большинство людей с синдромом Кляйнфельтера имеют мужской фенотип, нормальный интеллект, достаточный уровень потенции, они вступают в брак. Таким образом, показанием для исследования на половой хроматин является олигоазооспермия у пациента.
Синдром Ди Джорджи
При синдроме Ди Джорджи у больных отмечается наличие врожденной формы аплазии паращитовидных желез и тимуса. Является разновидностью идиопатического изолированного гипопаратиреоза. Встречается достаточно редко.
При этом заболевании патологические изменения касаются околощитовидных (паращитовидных) желез, у которых отмечается дисгенез или агенезия. Вилочковая железа (тимус) отсутствует от рождения. В результате сочетания таких патологий происходит резкое снижение числа Т-лимфоцитов, формируется иммунологическая недостаточность. Кроме того, этот синдром сопровождается формированием врожденных аномалий крупных сосудов.
Заболевание является аутосомным и определяется наличие мутации в 22 хромосоме. В большинстве случаев причиной является спорадическая делеция 22q11 (реже микроделеция 22q11.2). Наследование происходит по доминантному принципу, с полом не связано. Некоторые авторы не соглашаются с такой характеристикой и приводят аргументы в пользу аутосомно-рецессивного типа, обладающего различной эспрессивностью.
Для заболевания характерно нарушение процесса эмбриогенеза 3-4 жаберных карманов, что приводит к нарушению закладки вилочковой железы и паращитовидных желез.
В клинике наиболее постоянными симптомами являются кандидомикоз и гипопаратиреоз, довольно часто сопровождающиеся нарушением процесса формирования рта, носа и ушей.
Тимус из-за нарушения развития в эмбриональном периоде остается неразвитым. Эпителий тимуса не обеспечивает нормального процесса развития Т-клеток. В итоге формируется специфическая форма иммунодефицита, при которой ослабляется гуморальный иммунный ответ и ответ на клеточном уровне. Если у ребенка имеется подобное патологическое нарушение иммунитета, то он будет обладать повышенной чувствительностью к инфекциям бактериального, вирусного и грибкового происхождения.
Синдром может протекать в форме генетически обусловленного отсутствия паращитовидных желез или изолированной недостаточности околощитовидных желез – в сопровождении гипокальциемических судорог, которые начинаются от рождения. Иммунологическая недостаточность приводит к появлению различных инфекционных заболеваний. Как правило, совокупность симптомов вызывает сердечную недостаточность. Кроме того, летальный исход вызывают инфекционные болезни.
Диагностика синдрома предполагает выявление типичных для синдрома патологий: искажения формы лица и черепа, наличие иммунологической недостаточности, аплазии тимуса, дисгенезии или агенезии паращитовидных желез. Ярче всего при заболевании проявляются кандидомикоз и гипопаратиреоз.
Регуляция пола
В генетике человека она осуществляется двумя правилами: первое определяет зависимость развития зачаточных половых желез от секреции тестостерона и гормона MIS. Второе правило указывает на исключительную роль, которую играет У-хромосома. Мужской пол и все соответствующие ему анатомические и физиологические признаки развиваются под воздействием генов, находящихся в У-хромосоме. Взаимосвязь и зависимость обоих правил в генетике человека называется принципом роста: у эмбриона, являющегося бисексуальным (то есть имеющим зачатки женских желез – мюллерова протока и мужских гонад – вольфова канала) дифференцировка эмбриональной половой железы зависит от наличия или отсутствия в кариотипе У-хромосомы.
Аниридия
При аниридии нарушается нормальное строение глаза: в органе зрения отсутствует радужная оболочка. Кроме того, часто развиваются сопутствующие патологические изменения, такие как макулярная гипоплазия и гипоплазия зрительного нерва, изменения роговицы, катаракта. Острота зрения заметно падает, попытки коррекции не приносят существенных результатов. Развивается светобоязнь и горизонтальный нистагм. В некоторых случаях отмечается появление врожденной глаукомы.
Причиной заболевания является нарушение функционирования гена PAX6 из короткого плеча 11 хромосомы. Кодируемый им белок приводит к запуску ряда процессов, которые управляют процессом правильного формирования органов зрения и ряда других структур. Примечательно, что ген очень консервативен: отличие форм PAX6 у человека и данио рерио составляет менее 5%, несмотря на расхождение эволюционных линий примерно 400 млн лет назад.
Заболевание относится к группе аутосомно-доминантных патологий. В случае гомозиготности по мутантной копии гена PAX6 негативный эффект на организм возрастает, что вызывает множественные нарушения в работе органов зрения. Кроме того, поражается ЦНС, что приводит к летальному исходу.
Лечение направлено на сглаживание симптомов. Для визуальной имитации зрачка рекомендуется использовать специальным образом окрашенные линзы. Возможно восстановление зрачка путем реконструктивной пластической операции.
Половые хромосомы у цветковых растений
Для большинства цветущих растений или покрытосеменных растений мужские и женские половые органы присутствуют на одном и том же цветке. Иногда один растение может производить отдельные мужские и женские цветы для усиления креста оплодотворение или мужские и женские половые органы могут созревать в разное время. Тем не менее, присутствие различных мужских и женских растений встречается относительно редко, и только шесть процентов покрытосеменных растений показывают эту характеристику, которая называется диоэксизмом. Даже те, у кого этот вид полового диморфизма возрастает из-за стерильных мужских или женских стерильных мутаций и, следовательно, различных половых хромосом, известны только в четырех семействах растений.
Похоже, что растения находятся на ранних стадиях эволюции гетероморфных половых хромосом. Поэтому они могут быть использованы в качестве моделей для изучения событий, которые приводят к определению хромосомного пола.
Количество в различных организмах
У эукариот
В этих таблицах указано общее количество хромосом (включая половые хромосомы) в ядре клетки. Например, большинство эукариот имеют диплоидный , как , у которых есть 22 различных типов аутосом , каждый из присутствующих в виде двух гомологичных пар, и две половые хромосомы . Всего получается 46 хромосом. Другие организмы имеют более двух копий своих типов хромосом, например, мягкая пшеница , которая является гексаплоидной и имеет шесть копий семи различных типов хромосом — всего 42 хромосомы.
|
|
|
Нормальные представители определенного вида эукариот имеют одинаковое количество ядерных хромосом (см. Таблицу). Другие эукариотические хромосомы, то есть митохондриальные и плазмидоподобные маленькие хромосомы, гораздо более разнообразны по количеству, и могут быть тысячи копий на клетку.
23 хромосомные территории человека во время прометафазы в клетках фибробластов
Виды, размножающиеся бесполым путем, имеют один набор хромосом, одинаковый во всех клетках организма. Однако бесполые виды могут быть гаплоидными или диплоидными.
У видов, размножающихся половым путем, есть соматические клетки ( клетки тела), которые являются диплоидными , имеющими два набора хромосом (23 пары у человека), один набор от матери и один от отца. Гаметы , репродуктивные клетки, гаплоидны : они имеют один набор хромосом. Гаметы продуцируются мейозом диплоидной клетки зародышевой линии . Во время мейоза совпадающие хромосомы отца и матери могут обмениваться небольшими частями самих себя ( кроссовер ) и, таким образом, создавать новые хромосомы, которые не наследуются исключительно от одного из родителей. Когда мужская и женская гамета сливаются ( оплодотворение ), образуется новый диплоидный организм.
Некоторые виды животных и растений являются полиплоидными : они имеют более двух наборов гомологичных хромосом . Растения, важные для сельского хозяйства, такие как табак или пшеница, часто полиплоидны по сравнению с их предковыми видами. Пшеница имеет гаплоидное число из семи хромосом, которое до сих пор встречается у некоторых сортов, а также у диких предков. Более распространенные типы макаронных изделий и мягкой пшеницы являются полиплоидными с 28 (тетраплоидными) и 42 (гексаплоидными) хромосомами по сравнению с 14 (диплоидными) хромосомами у дикой пшеницы.
У прокариот
Виды прокариот обычно имеют по одной копии каждой основной хромосомы, но большинство клеток могут легко выжить с несколькими копиями. Например, Buchnera , A симбионт из тлей имеет несколько копий своей хромосомы, в пределах от 10-400 копий на клетку. Однако у некоторых крупных бактерий, таких как Epulopiscium fishelsoni, может присутствовать до 100 000 копий хромосомы. Плазмиды и маленькие плазмидоподобные хромосомы, как и у эукариот, сильно различаются по количеству копий. Количество плазмид в клетке почти полностью определяется скоростью деления плазмиды — быстрое деление вызывает большое количество копий.