Болезнь (синдром) марфана
Содержание:
Причины синдрома Марфана
Синдром Марфана – генетическое заболевание с наследуемостью аутосомно-доминантного типа, причиной его развития являются мутации гена FBN1
Этот ген отвечает за выработку фибриллина-1 – важного структурного белка, входящего в состав связок и эластичных сосудов. При нарушениях в строении этого белка соединительнотканные структуры становятся более растяжимыми, при этом теряют устойчивость к деформациям. Наиболее опасные последствия таких изменений проявляются со стороны органов зрения и сердечнососудистой системы
Фибриллин-1 необходим для нормального функционирования цинновой связки, прикрепляющей хрусталик глаза к цилиарному телу. Поэтому при нарушении его синтеза, что происходит у больных с синдромом Марфана, цинновая связка ослабляется, что на первых этапах проявляется близорукостью у пациента, подвывихом хрусталика, но в дальнейшем может привести к вторичной глаукоме, частичной или полной потере зрения
Наиболее опасные последствия таких изменений проявляются со стороны органов зрения и сердечнососудистой системы. Фибриллин-1 необходим для нормального функционирования цинновой связки, прикрепляющей хрусталик глаза к цилиарному телу. Поэтому при нарушении его синтеза, что происходит у больных с синдромом Марфана, цинновая связка ослабляется, что на первых этапах проявляется близорукостью у пациента, подвывихом хрусталика, но в дальнейшем может привести к вторичной глаукоме, частичной или полной потере зрения.
Другие функции фибриллина в организме – формирование внеклеточного матрикса, что обеспечивает целостность соединительных тканей и нормальное функционирование факторов роста клеток.
Аорта относится к сосудам эластического типа и содержит множество эластических связок, от которых зависит ее устойчивость к нагрузкам. При нарушении их функции возникает расширение аорты, расслоение ее стенок. Такая ситуация ставит под угрозу жизнь пациента, может спровоцировать внезапную смертность из-за разрыва аорты во время интенсивных физических нагрузок. Расширение аорты опасно для женщин в период беременности, особенно это касается третьего триместра, а также во время родов и несколько месяцев после них.
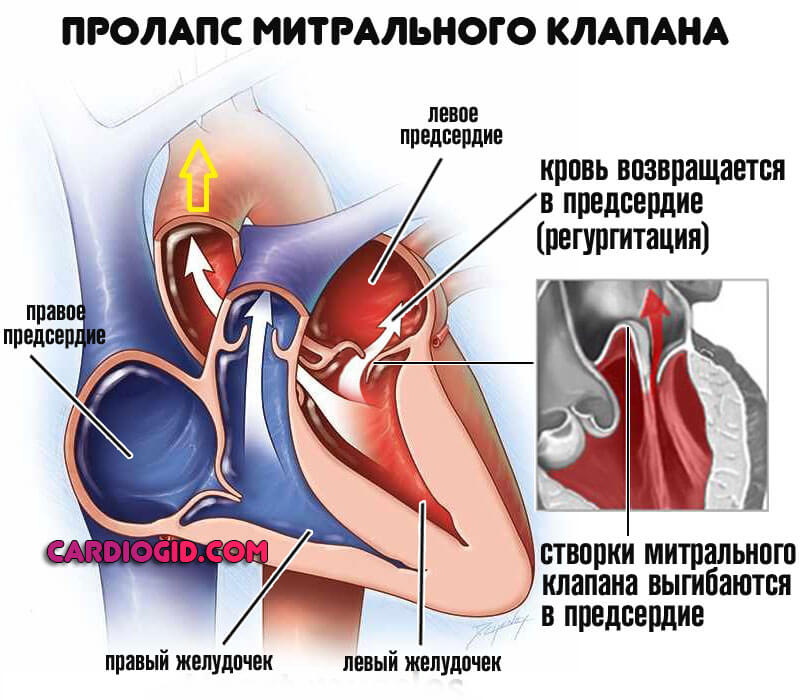
Среди других распространенных сердечных патологий, связанных с синдромом Марфана – поражения митрального клапана, из-за чего возникает необходимость хирургического лечения.
Синдром Марфана наследуется?
Синдром Марфана – генетическое заболевание, которое в 75% случаях наследуется по семейному типу, а оставшиеся 25% встречаемости являются результатом первичной мутации гена. В семьях, где отец старше 35 лет риск рождения ребенка с синдромом Марфана повышается.
Развитие симптомов начинается уже в период перинатального развития – соединительнотканные волокна в процессе формирования теряют прочность, с чего начинаются изменения в эластических сосудах, опорно-двигательном аппарате, глазных связках, твердом небе, сердечных клапанах. Прогрессирование заболевания продолжается в постнатальный период, и длится всю жизнь пациента.
Методы терапии
Прежде всего, лечение заключается:
- в назначении бета-адреноблокаторов;
- в хирургическом вмешательстве при патологиях клапанов и аорты;
- в хирургической коррекции патологий позвоночника.
Из бета-блокаторов пациентам целесообразно назначать препараты в виде пропранолола или атенолола. Это помогает предотвратить серьёзные осложнения со стороны сосудов и сердца. Бета-блокаторы уменьшают интенсивность сокращения сердечной мышцы, снижая её нагрузку, останавливают процесс расслоения аорты и снижают риск развития аневризмы. Если аневризма достигает критических размеров, пациентам показано хирургическое вмешательство.
В качестве консервативного лечения сколиоза, обычно, применяют фиксацию позвоночника, но если он искривлён от 40 градусов и более, операция является более предпочтительным методом.
Всем пациентам, страдающим синдромом Марфана, нужно каждый год проходить обследование у невролога, кардиолога и окулиста, с генетическим консультированием по показаниям.
Лечение синдрома Марфана
От синдрома Марфана полностью избавиться и устранить механизм его развития невозможно. Лечение базируется на улучшении общего состояния больного, устранении клинических проявлений и проведении профилактических мероприятий, препятствующих развитию осложнений.
Больным с данным синдромам рекомендовано ограничить физическую нагрузку до низкого уровня, или минимального. Риск появления патологии сердечно-сосудистой системы возрастает при средних и высоких физических нагрузках.
Следует обходить стороной и повседневные нагрузки, при которых возможно повышение внутригрудного давления, ведущему к развитию пневмоторакса (например, подъем тяжестей, подъем по этажам).
Медикаментозное лечение
Медикаментозная терапия направлена на устранение клинической картины заболевания.
Со стороны сердечно-сосудистой системы рекомендованы β-адреноблокаторы (например: Анаприлин), которые снижают скорость распространения пульсовых волн при стремительно растущем расширении аорты и обратного тока крови на двустворчатом клапане или клапане аорты.
β-адреноблокаторы также оказывают положительный эффект при нарушении ритма и проводимости, в сочетании с сердечными гликозидами.
Но следует помнить о существующих противопоказаниях данных групп препаратов:
- хронический обструктивный бронхит;
- бронхиальная астма;
- снижение частоты сердечных сокращений;
- низкое артериальное давление.
Блокаторы каналов кальция используются при наличии противопоказаний к В-адреноблокаторам.
Хирургия
Хирургическое лечение проводится, если есть осложнения со стороны сердечно-сосудистой системы, с целью коррекции пораженных участков. Его проводят при пролабировании двустворчатого клапана и расслаивании аорты.
При этом осуществляется протезирование двустворчатого клапана.
У беременных с тяжелым течением болезни Марфана, роды разрешаются хирургическим путем.
Генетика
Генетичний характер СМ вперше помітив H. Weve, який описав родину з кількома хворими і таким чином довів аутосомно-домінантний тип успадкування (Лисиченко О.В., 1986). Результати подальших генетичних досліджень свідчать, що мутація в гені FBN1 виявляється не лише у пацієнтів із СМ, але й при інших подібних захворюваннях сполучної тканини, що об’єднані в групу фібрилінопатій І типу (Фищенко Я.В., 2006). Ген FBN1 розміщується на довгому плечі 15-ї хромосоми і картований в локусі 15q 21.1 (рисунок).
Рисунок
Структура 15-ї хромосоми людини
15-та хромосома людини — одна з 23 пар людських хромосом, яка містить >102 млн пар основ, що становить 3–3,5% усього матеріалу ДНК клітини людини. Ймовірно, вона містить 700–900 генів
Приблизно у 75% випадків захворювання передається генетично і лише 25% викликаються спорадичними мутаціями. Варто зазначити, що СМ характеризується вираженою генетичною гетерогенністю. Сьогодні відомо близько 550 мутацій у різних родинах. Серед виражених мутацій в гені FBN1: 57% — міссенс-мутації, 18% — фреймшифт-мутації, 16% — спайс-сайт, 8% — нонсенс-мутації. Для класичної форми СМ характерна мутація в одному із доменів FBN1 (epidermal growth factor (EGF)-like domain), що відповідає за зв’язування кальцію з фібриліном. Патологічні зміни в тому самому локусі можуть викликати різні клінічні прояви — від стертої форми з ураженням однієї системи організму до класичної форми. Клінічна різноманітність СМ зумовлює втягнення мутацій, локалізованих в інших генах, наприклад в гені FBN2 або FBN3. Цей факт підтверджується тим, що у частини пацієнтів із клінічно вираженим СМ визначається нормальний метаболізм фібриліну, а при генетичному аналізі відсутня мутація в гені FBN1 (Ватутин Н.Т. и соавт., 2006; Фищенко Я.В., 2006).
Вступ
Синдром Марфана (СМ) — захворювання сполучної тканини з ураженням скелетно-м’язової, серцево-судинної систем та очей, яке успадковується за аутосомно-домінантним типом. Незважаючи на те що СМ був описаний більше як 100 років тому, він і надалі залишається однією з актуальних проблем медицини з позиції етіології, патогенезу, морфогенезу, діагностики та лікування. У зв’язку з цим і досі переглядаються діагностичні критерії, класифікація СМ, вивчаються фенотипові прояви захворювання та ведуться пошуки нових методів лікування. В одного пацієнта із СМ може бути стільки проблем зі здоров’ям, скільки є спеціалістів у поліклініці.
Лечение
Терапия представляет собой хирургическую и консервативную коррекцию сердечнососудистых патологий.
В основном, лечебные меры нацелены на профилактику развития недуга и осложнений. Больным назначают антагонисты кальция, ингибиторы АПФ и другие препараты.
По показаниям делается операция при пролапсе, недостаточности и других нарушениях.
Необходимо обязательно заниматься коррекцией зрения. Для этого пациентам рекомендованы очки и линзы, иногда прибегают к лазерному воздействию.
При скелетных расстройствах выполняют стабилизацию позвоночника хирургическим путем.
Помимо всего прочего лечебный курс состоит из приема витаминов и метаболического лечения.
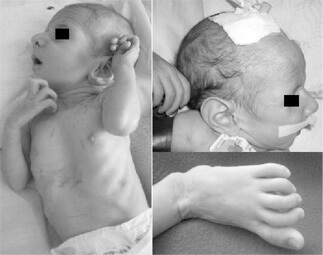
Синдром марфана у детей можно легко распознать по характерным признакам, описанным выше. Неестественно длинные пальцы видны уже у новорожденных, что должно послужить поводом к обращению в медицинское учреждение.
При раннем половом созревании проводится индукция специальными гормональными препаратами. В остальном, тактика лечения точно такая же, как и у взрослых.
Пациентам обязательно нужно ходить в ортопедической обуви.
Синдром марфана и беременность — это несовместимые вещи. Заболевание относят к числу очень редких, потому при вынашивании недуг может приводить к трагическим последствиям. Довольно часто возникают выкидыши и преждевременные роды с таким заболеванием. Были даже смертельные случаи.
Прогноз при беременности крайне неблагоприятен. Это обусловлено такими причинами:
- Во-первых, появляется риск развития недуга у ребенка.
- Во-вторых, существует риск осложнений в виде аневризмы аорты и септического эндокардита.
Історична довідка
У 1896 р. французький педіатр Антуан Жан Марфан описав 5-річну дівчинку із надзвичайно тонкими і довгими кінцівками, контрактурами суглобів, кіфосколіозом і назвав цей синдром pattes d’araignee (пальці павука). Ця дівчинка могла мати арахнодактилію з контрактурами (синдром Білса), але ім’я Марфана було використано для позначення поєднання симптомів, пов’язаних із дефектами в гені фібриліну-1 (fibrillin-1 — FBN1). Пізніше терапевт Е.C. Achard (1902) назвав його арахнодактилією і доліхостеномелією (від грец. dolicos — довгі, stenos — тонкі, melis — кінцівки) (Лисиченко О.В., 1986). Існують інші назви, що нині становлять лише історичний інтерес: синдром Марфана — Ешера; акромакрія Пфаундлера; акродоліхія Бругшмеллера; доліхостеномелія; синдром Марфана — Ербе; гіперхондроплазія Мері та Бабоне; акрохондрогіперплазія Валентина; вроджена акромакрія з аміотонією Юнга; вроджена мезодермальна дистрофія Веве; дисмезектопія Лавал; доліхоморфія. Окрім цього, частина авторів вважає, що сучасне визначення синдрому Марфана не має жодного відношення до описаного Марфаном у 1896 р. (Лазовскис И.Р., 1981).
Ураження аорти як симптом СМ виявлено лише у 1943 р. L.E. Etter та L.P. Glover, а роль дилатації аорти в скороченні тривалості життя уточнено у 1972 р. J.L. Murdoch. У 1990 р. E.T. Холлістером описана роль фібриліну в патогенезі СМ, а також визначений локус цього захворювання у хромосомі 15q21.1. Доказ того, що мутації гена FBN1 можуть призвести до СМ, описані у 1991 р. Даєцом (цит. за: Frydman M., 2008).
Синдром Марфана — рідкісне захворювання з частотою виникнення 2–3 випадки на 10 тис. осіб (Барашнев Ю.И. и соавт., 1983). Більшість авторів описували поодинокі випадки захворювання. За даними О.В. Лисиченка (1986), найбільша кількість випадків описана у Марфана (1938) — 10, Грімальді (1964) — 15, Макк’юсика (1966) — 75 пробандів. Вітчизняні дослідники Кондрашин, Неудахін (1968) діагностували синдром Марфана у 17; Надарейшвілі, Паламарчук (1970) — у 11; Ласкова, Ласков (1970) — у 19; Гусева (1971) — у 30; Лисиченко (1971) — у 64; Уткін, Грушин (1972) — у 23; Гапузов (1973) — у 10; Сємячкіна (1975) — у 70 хворих.
Прогноз
До появления современных сердечно-сосудистых хирургических методов и лекарств, таких как лозартан и метопролол , прогноз для пациентов с синдромом Марфана был неблагоприятным: ряд неизлечимых сердечно-сосудистых заболеваний был обычным явлением. Продолжительность жизни сократилась как минимум на треть, и многие умерли в подростковом и двадцатилетнем возрасте из-за сердечно-сосудистых заболеваний
Сегодня сердечно-сосудистые симптомы синдрома Марфана по-прежнему являются наиболее важной проблемой в диагностике и лечении заболевания, но адекватный профилактический мониторинг и профилактическая терапия предлагают что-то, приближающееся к нормальной продолжительности жизни, и по мере того, как все больше пациентов живут дольше, обнаруживается все больше проявлений болезни. Женщины с синдромом Марфана живут дольше мужчин.
Симптоматика со стороны разных органов и систем
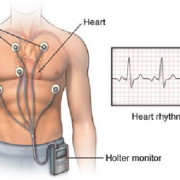
Со стороны сердца и сосудов у больных чаще всего выявляют пролапс клапана и аортальную аневризму. Если корень аорты и её восходящая часть патологически изменены, это чревато серьёзными осложнениями. Как правило, повреждение интимы аорты происходит там, где нагрузка на неё особенно велика. Происходит постепенное расширение сосуда либо коронарный синус может внезапно разорваться.
Расширение корня аорты диагностируют у половины пациентов в детском возрасте и примерно в 60-80% случаев у взрослых. Аортальную недостаточность выявляют с помощью аускультации, в виде диастолического шума, прослушиваемого над клапаном. При растяжении клапанных створок и хорд может возникнуть пролапс митрального клапана с систолическим щелчком и шумом разной интенсивности. Не исключена возможность развития эндокардитов бактериальной природы как на аортальном, так и на митральном клапане.
Со стороны опорно-двигательной системы важным признаком является высокий рост пациента, с размахом рук, значительно превышающим их высоту. При более тщательном осмотре можно выявить арахнодактилию большого пальца с выступлением его дистальной фаланги за край руки, сжатой в кулак. Грудная клетка имеет форму киля со смещением наружу либо воронки — со смещением внутрь. Суставы часто гиперподвижны, с незначительными контрактурами в области локтей. Колени могут быть выгнуты назад, нередко плоскостопие и искривление позвоночника по типу кифоза и сколиоза. Также наблюдается слабое развитие подкожно-жировой клетчатки.
Эктопия хрусталика (его подвывих либо он смещён вверх) является основным признаком синдрома Марфана со стороны зрительной системы. Также типичны явления иридоденеза, когда радужная оболочка глаз дрожит. Если поражён хрусталик, край его дислоцирован, что можно увидеть, когда зрачки пациента не расширены. Близорукость со спонтанным отслоением глазной сетчатки тоже нередки.
Органы дыхания часто дают патологическую симптоматику в виде кистозной лёгочной болезни и спонтанного пневмоторакса, склонного к постоянному рецидивированию. В результате пациентов часто беспокоят одышка и боли в грудной клетке.
Что касается симптомов со стороны нервной системы, обследование показывает, что дуральный мешок, который окружает спинной мозг, сильно расширен в области поясницы и крестца. Это вызывает боли как в голове, так и в спине, а если, при этом, имеется неврологическая патология, функции мочеиспускания и дефекации могут быть значительно нарушены.
Что такое Болезнь (синдром) Марфана —
Болезнь Марфана (синдром Марфана) – это врожденная коллагенопатия или системная недостаточность соединительной ткани, для которой характерный определенный тип наследования и выраженной клинической симптоматики: проявления скелетной, сердечно-сосудистой и глазной патологии. Больные с синдромом Марфана имеют в своем анамнезе аневризмы аорты, гигантизм, миопию, долихостеномелию и арахнодактилию, деформацию грудной клетки, протрузию вертлужной впадины, эктопию хрусталика, плоскостопие, эктазию твердой мозговой оболочки, кифосколиоз.
Заболевание получило название в честь французского педиатра Антуана Марфана, который впервые в 1886 г. описал пятилетнюю девочку, имеющую длинные тонкие ноги и «паучьи» пальцы.
Соотношение распространенности данного врожденного заболевания составляет примерно 1:10 000 людей и мало зависит от пола. Однако процентный показатель среди детей и подростков достигает 6,8%, отметим, что большую часть из них формируют мальчики.
Клиническая картина
Формы течения заболевания:
- Стертая: выявляются минимальные органные изменения, охватывающие одну или две системы организма.
- Выраженная:
- минимальные патологические нарушения, охватывающие три системы организма;
- значительные патологические нарушения, которые возникают, как правило, в одной системе организма;
- значительные патологические нарушения, охватывающие две и более системы организма.
Заболевание может характеризоваться стабильным или прогрессирующим течением. В зависимости от генетической характеристики может иметь наследственный характер как впервые возникшая генная мутация.
Термин «болезнь Марфана» применяется при выявлении не менее трех классических проявлений заболевания и семейного характера заболевания.
Термин «синдром Марфана» обычно подразумевает стертое течение заболевания с положительными далее перечисленной клинической симптоматикой и органными изменениями.
«Марфаноподобный синдром», который нередко выявляется сразу после рождения ребенка, отличается множеством вариаций неполного набора Гентских критериев.
Рост больных нередко выше среднего, выявляются арахнодактилия, нефизиологическая гипермобильность суставов.
Зачастую развиваются аномалии строения скелета: сколиоз, кифоз, межпозвонковые грыжи, воронкообразная или килевидная деформация передней области грудной клетки, долихоцефалия, узкая форма лица, аномалия развития неба (высокое готическое небо), плоскостопие, сандалевидная щель, арахнодактилия.
Характерными являются изменения в глазном яблоке: вывих или подвывих хрусталика, сферофакия, глаукома, катаракта, отслойка сетчатки, мегалокорнеа, миопия, голубое окрашивание склер.
Изменения мышечной системы проявляются грыжами передней брюшной стенки, мышечной слабостью, нефроптозом.
Со стороны сердечно-сосудистой системы, как правило, возникают патологические изменения аорты (в 65–100 % случаях) и структуры клапанов. Патологические изменения аорты характеризуются расширением в области синуса Вальсальвы и/или восходящей части дуги аорты. Изолированное расширение коня аорты обычно не сопровождается какой-либо клинической симптоматикой, иногда могут возникать загрудинные боли или чувство дискомфорта при физической нагрузке – аорталгии, развивающиеся от чрезмерного растяжения аортальной стенки.
Наряду с расширением синуса Вальсальвы и восходящего участка аорты, при болезни Марфана нередко формируется аневризма дуги, нисходящей части и брюшного отдела аорты. В зависимости от уровня возникновения аневризмы могут у пациента развиваются головные боли, отечность лица, боли за грудиной и/или в межлопаточной области, охриплость голоса (при возникновении сдавления возвратного нерва), рефлекторный кашель, нарушения при глотании. Больные могут предъявлять жалобы на боли в эпигастральной области, чувство тяжести и усиленную пульсацию в животе.
При разрыве аневризмы аорты развивается сердечно-сосудистый коллапс и шок, зачастую с летальным исходом.
Помимо аневризмы аорты, при синдроме Марфана часто наблюдается патология клапанного аппарата сердца, наиболее часто – пролапс митрального клапана.
Поражение органов дыхания при синдроме Марфана диагностируется в 10–25 % случаев и объясняется нарушением структурных элементов легочной ткани в виде полного или частичного разрушения межальвеолярных перегородок, недоразвитием эластических мышечных волокон в терминальных бронхах и бронхиолах, что становится причиной повышения растяжимости и снижения эластичности легочной ткани, развития апикальной или диффузной буллезной эмфиземы.
Вовлечение в патологический процесс других внутренних органов характеризуется возникновением висцероптоза (птоз желудка, мочевого пузыря, маки), недостаточностью кардиального отдела желудка, эктазией твердой мозговой оболочки в пояснично-крестцовом отделе. В почках возможно возникновение аплазии и поликистоза.
2.Наследственные заболевания соединительной ткани
Некоторые болезни соединительной ткани являются результатом изменений в определенных генах. Вот наиболее распространенные из них:
Синдром Элерса-Данло
Данный синдром представляет собой группу заболеваний наследственного характера, отличительной особенностью которых являются гипермягкие суставы, повышенная эластичность кожи, подверженные повреждениям кровеносные сосуды, аномальный рост рубцовой ткани и синяки. Симптоматика данного синдрома может варьироваться от легкой степени до высокой. В зависимости от конкретного вида Элерса-Данло признаки этого заболевания могут включать следующее:
- кровоточивость десен,
- слабые сосуды,
- медленное заживление ран,
- изогнутый позвоночник,
- плоскостопие,
- проблемы с легкими, сердечными клапанами и органами пищеварения.
Врожденный буллезный эпидермолиз
Люди с данным заболеванием имеют настолько хрупкую кожу, что практически любое соприкосновение с ней сопровождается возникновением тонкостенных волдырей с серозным содержимым. Подобные пузыри могут возникнуть при ударе, падении человека на землю и даже от трения одежды на определенных участках кожи.
В зависимости от какого-либо конкретного вида врожденного буллезного эпидермолиза подвергаться неблагоприятному воздействию могут дыхательные пути, пищеварительный тракт, мочевой пузырь, мышцы или другие органы. Как правило, буллезный эпидермолиз проявляется сразу при рождении ребенка, так как сам акт родов в некоторой степени является первой механической травмой.
Синдром Марфана
Клиническая картина данного заболевания характеризуется поражением большинства жизненно важных органов и систем: сердечно-сосудистой и центральной нервной системы, опорно-двигательного аппарата, органов зрения и дыхания. Люди с синдромом Марфана имеет высокий рост, а также чрезмерно длинные кости и тонкие «паукообразные» пальцы ног, рук.
Среди других проблем, сопровождающих синдром Марфана, можно выделить проблемы со зрением по причине аномального размещения хрусталика глаза и расширения аорты. Синдром Марфана провоцируется мутациями в гене, который регулирует структуру белка фибриллина.
Несовершенный остеогенез
Данная патология является врожденным расстройством, характеризующимся хрупкостью костей, низкой мышечной массой. Существует несколько типов этого заболевания. Специфические симптомы несовершенного остеогенеза зависят от конкретного вида заболевания и могут включать в себя следующие признаки:
- серый или синий оттенок склеры – белка глаз;
- тонкая кожа;
- снижение слуха;
- незначительное искривление позвоночника;
- проблемы с дыханием.
Причина возникновения данной патологии заключается в мутации генов COL1A1 и COL1A2, ответственных за коллаген типа 1, а также изменение качества протеина.