Потребление нутриентов при тренировке с отягощениями
Содержание:
- Убихинон
- Экзогенный путь метаболизма липидов
- Общий принцип биосинтеза липидов
- Биологическая роль селен-цистеин-содержащей тРНК и селенсодержащих протеинов
- Гистохимические методы определения в тканях
- Биосинтез холестерина
- Регуляция плазменного содержания апо-B-липопротеина в норме и при нарушениях липидного обмена
- Обратный транспорт ХС
- Эмульгирование и гидролиз липидов
- Биохимические методы исследования
Убихинон
Убихинон (коэнзим Q10) состоит из гексамерного хинольного кольца (Q) и десяти остатков цепи молекулы изопрена. Он принимает участие как постоянный компонент цикла Кребса в акцепции окисленных остатков от фиксированных флавоноидных комплексов и транспортирует их далее по цитохромам дыхательной цепи митохондрий.
Таким образом, ХС является структурным компонентом биологических мембран, участвует в процессах биосинтеза ряда витаминов, стероидных гормонов, желчных кислот, а также коферментов, играющих важную роль в регуляции процессов пластического и метаболического обмена веществ.
Экзогенный путь метаболизма липидов
Более 95% липидов, поступающих с пищей, представлены ТГ, остальное количество составляют фосфолипиды, СЖК, ХС, который присутствует в пищевых продуктах преимущественно в виде эстерифицированного ХС, а также жирорастворимые витамины. В желудке и двенадцатиперстной кишке под влиянием солей желчных кислот осуществляется процесс эмульгирования липидов (рис. 2.1). Натриевые соли желчных кислот, располагаясь на поверхности капелек липидов, снижают их поверхностное натяжение и способствуют дисперсии последних, увеличивая их суммарную поверхность. Образующаяся тонкодисперсная эмульсия липидов в воде облегчает их взаимодействие с панкреатической липазой. Под действием последней ТГ превращаются в моноглицериды и СЖК. Эфиры ХС, содержащиеся в пище, подвергаются деэстерификации в свободный ХС также под влиянием липаз. Моноглицериды, СЖК и свободный ХС эмульгируются желчными кислотами и затем абсорбируются энтероцитами, после чего конденсируются с ТГ и вместе с ХС включаются в состав хиломикронов. Кроме того, моноглицериды и СЖК вместе с солями желчных кислот в тонком кишечнике образуют мицеллы, размеры которых сопоставимы с размерами молекул липидов. Мицеллы всасываются энтероцитами тонкого кишечника посредством эндоцитоза. В энтероцитах осуществляется ресинтез ТГ из моноглицеридов и СЖК мицелл. Ресинтезированные ТГ энтероцитарного происхождения в дальнейшем инкорпорируются в хиломикроны. Таким образом, хиломикроны являются основной транспортной формой экзогенных ТГ, осуществляя их трансфер из энтероцитов тонкого кишечника в системный кровоток через воротную вену и грудной лимфатический проток. В качестве главного структурного протеина хиломикроны содержат апо-В48-протеин, а период их полужизни обычно не превышает 5–20 мин. Необходимо отметить, что плазма крови здоровых людей при взятии крови натощак практически не содержит хиломикронов.
После секреции хиломикроны получают аполипопротеины классов Е, С-I, C-II и C-III от ЛПВП. В плазме крови апо-C-II-липопротеин активирует эндотелиальную липопротеинлипазу. Под действием последней большинство ТГ в хиломикронах расщепляется до глицерина и свободных НЭЖК. Белково-липидный комплекс, ответственный за их транспорт, отличается очень высокой плотностью и малыми размерами. НЭЖК используются в жировой и мышечной ткани в качестве энергетического субстрата. В то же время хиломикроны подвергаются так называемому метаболическому ремоделированию с образованием остатков (ремнантов) с более низкой плотностью: ЛПОНП и ЛППП. Ремнанты хиломикронов, содержащие апо-В48-липопротеин и обогащенные апо-Е-липопротеином, захватываются гепатоцитами с помощью специфических рецепторов, имеющих высокое сродство к последнему. В дальнейшем ТГ, ассоциированные с ремнантами хиломикронов, подвергаются воздействию внутрипеченочной липопротеинлипазы. В результате ресинтезируются СЖК и глицерин, которые используются для продукции липидов, либо вовлекаются в процессы β-окисления.
Желчные кислоты, всасывающиеся в тонком кишечнике, по системе воротной вены транспортируются в гепатоциты, а затем секретируются в желчные капилляры вместе с ресинтезированными желчными кислотами. Подобная внутрипеченочная рециркуляция предотвращает экзогенные потери желчных кислот и поддерживает синтез ХС в гепатоцитах на относительно стабильном уровне.
Экзогенный ХС в ограниченном количестве (не более 0,5 г/сут) всасывается в кишечнике в составе липидных мицелл. Далее он в составе хиломикронов достигает гепатоцитов, где подвергается эстерификации, либо превращается в соли желчных кислот. Причем интенсивность секреции ХС в желчный капилляр и ретенция в гепатоците, регулируемая специфическими молекулами ABCG5 и ABCG8 (ATP (adenosine triphosphate)-binding cassette (ABC) transporter G5 и G8 — АТФ (аденозинтрифосфат)-связанный кассетный переносчик G5 и G8), являются взаимо взвешенными процессами (Yu L. et al., 2002).
Биосинтез эндогенного ХС из ацетил-КоА осуществляется в основном в гепатоцитах и в следовых количествах — в энтероцитах тонкого кишечника. Часть ХС выводится с желчью, а другая — подвергается эстерификации.
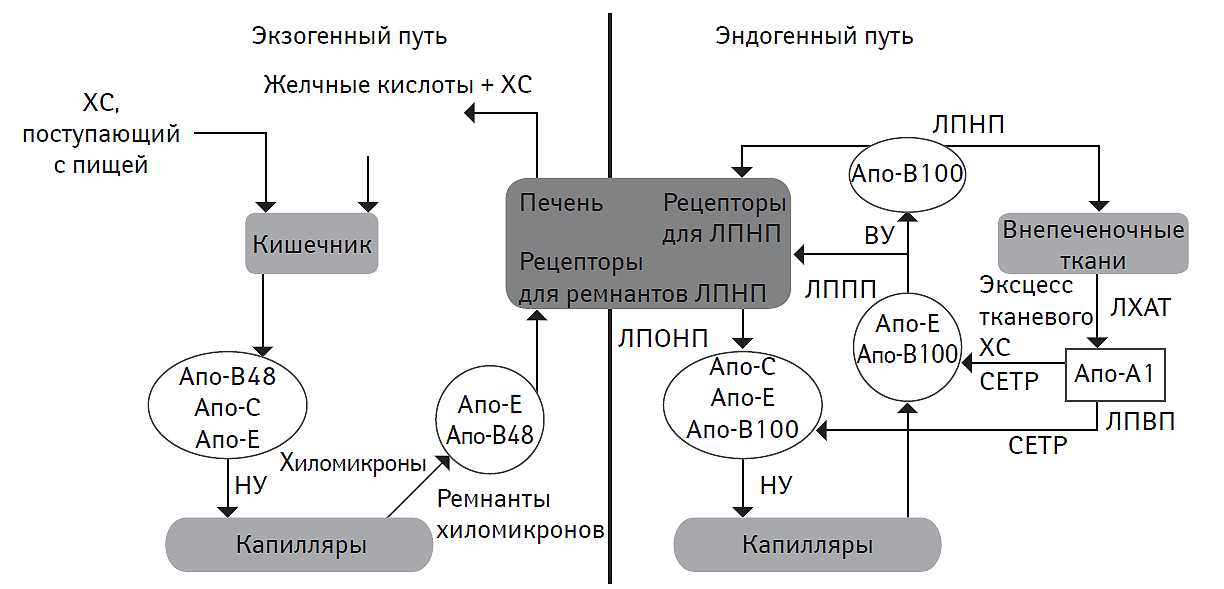
Рис. 2.1. Основные пути метаболизма липидов
На рис. 2.1, 2.2 и 2.3: ЛХАТ — лецитин-холестерин-ацетилтрансфераза, CETP (cholesteryl ester transfer protein) — протеин, транспортирующий эстерифицированный ХС, НУ – низкий уровень, ВУ — высокий уровень.
Общий принцип биосинтеза липидов
Образование ЖК и их производных начинается с цитоплазмы. Вторая часть биосинтеза – удлинение молекулярной цепи также продолжается в клетке, однако «производственная мастерская смещается» внутрь митохондрии. На каждом этапе, соединение обогащается двумя атомами C, что напоминает процесс бета-окисления, только в его обратной интерпретации.
Говоря более развернуто, в цитоплазме непосредственно и происходит синтез, например пальмитиновой кислоты. Митохондрии же, используют уже готовый «полуфабрикат», для производства полноценных жирных кислот, состоящих из 18-и и более атомов углерода. Выполнить весь биосинтез самостоятельно от «А» до «Я», митохондрии не в состоянии. Причина банальна – «низкий уровень квалификации». Возвращаясь к технической терминологии, митохондрии обладают очень низкой способностью включать меченые уксусные кислоты в длинную цепь липидных структур.
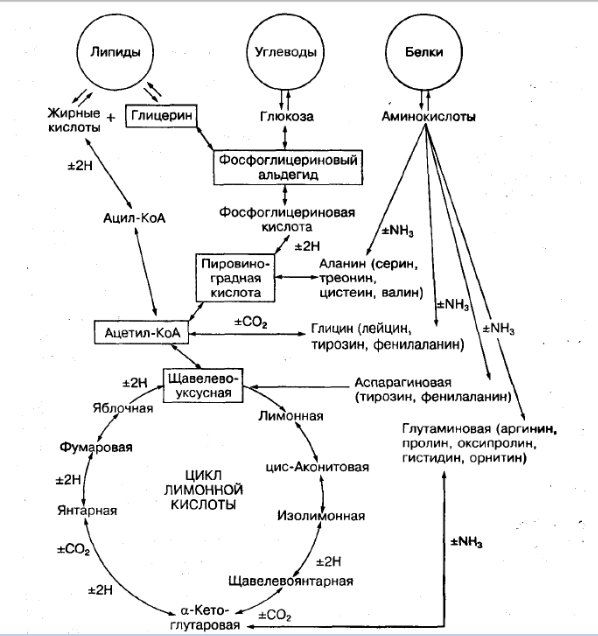
Биологическая роль селен-цистеин-содержащей тРНК и селенсодержащих протеинов
Селен-цистеин-содержащая тРНК является производным изопентенилированного аденозина-37 (A37), обычно опосредованного модификацией антикодона в положении 3′ (Moustafa M.E. et al., 2001). Селен-цистеин-содержащая тРНК декодирует UGA, который в физиологических условиях блокирует кодон и обеспечивает включение молекулы селен-цистеин в формирующиеся селенсодержащие пептиды. Отсутствие изопентенилированной тРНК проявляется в снижении эффективности деградации селен-цистеин-содержащей тРНК в декодированных нечувствительных кодонах бактериальных и дрожжевых клеток (Warner G.J. et al., 2000). Таким образом, неточная трансляция сигнала остановки процесса кодирования может привести к преждевременному завершению процесса протеинового синтеза и сборке короткоцепочечных белковых молекул.
Гистохимические методы определения в тканях
Самым старым методом окрашивания Л. в тканях является метод с использованием четырехокиси осмия (OsO4). Этот реактив восстанавливается непредельными жирными к-тами и целым рядом других веществ, обладающих восстанавливающими свойствами. Продукты восстановления OsO4 окрашены в черный цвет. Однако следует признать, что методы выявления Л. с помощью жирорастворимых красителей более просты и надежны. В гистохимии для этих целей прежде всего стали использовать судан III, несколько позже — судан IV и шарлах. Л. более интенсивно окрашиваются красящими смесями, особенно теми, которые содержат два (или более) гомолога или изомера нафтоловых суданов. Окрашивание Л. жирорастворимыми красителями основано на том, что они растворяются в жировых веществах лучше, чем в обычных растворителях. Термин «суданофилия» означает способность ткани окрашиваться любыми жирорастворимыми красителями.
Для сохранения Л. в тканях при фиксации рекомендуется использовать 10 — 15% р-р формалина, но еще лучше использовать фиксатор формол-кальций по Бейкеру: формалин— 10 мл; 10% хлористый кальций — 10 мл; дистиллированная вода — 80 мл.
К этому фиксатору должен быть добавлен мел, для того чтобы смесь имела нейтральную реакцию. Фиксировать ткань рекомендуется 24—48 час., более длительная фиксация может привести к образованию кристаллов, изменению растворимости Л. и т. д. Отмытая после фиксации ткань промывается в проточной воде; срезы готовятся на замораживающем микротоме. Ткань паренхиматозных органов можно предварительно заключить в желатину.
При окрашивании ткани на Л. дает хорошие результаты и одновременно выявляет суданофильную зернистость в сегментоядерных лейкоцитах метод Гольдмана. Р-р судана III для окраски тканей по этому методу готовится следующим образом: 70% этанол — 100 мл; дистиллированная вода —- 20 мл; альфа-нафтол — 1,2 г; судан III — в избытке.
Смесь кипятят в течение 10 мин. и фильтруют. Срезы ткани красят 15 мин., затем дифференцируют в 70% этаноле, контролируя процесс под микроскопом. Мазки крови фиксируют 3 мин. смесью, состоящей из 1 части формалина и 4 частей 96 % этанола.
При окраске тканей на Л. по методу Чаччо следует маленькие кусочки фиксировать в течение 24—48 час. в смеси следующего состава: 5% водный р-р двухромовокислого калия — 80 мл; формалин — 20 мл; ледяная уксусная к-та — 5 мл.
Затем кусочки ткани выдерживают 5 — 8 дней в 3% двухромовокислом калии («хромируют»), сутки промывают в проточной воде, проводят через этанол восходящих концентраций в течение суток, проводят через ксилол и заключают в парафин. Приготовленные срезы после обработки 70% этанолом красят насыщенным р-ром судана III в 70% этаноле или при температуре 50° красителем следующего состава: 80 % этанол — 95 мл; ацетон — 5 мл; судан III — до насыщения.
После охлаждения жидкость фильтруется. Срезы красят 30 — 60 мин. при температуре 30°, споласкивают 50% этанолом, промывают в дистиллированной воде и заключают в глицерин-желатину.
Ядра клеток можно красить на Л. квасцовым гематоксилином, лучше это делать до обработки срезов су-даном. Л. окрашиваются в оранжевокрасный цвет.
Библиография: Алимова Е. К., Аствацатурьян А. Т. и Жаров Л. В. Липиды и жирные кислоты в норме и при ряде патологических состояний, М., 1975; Биохимические методы исследования в клинике, под ред. А. А. Покровского, М., 1969; Кейтс М. Техника липидологии, пер. с англ., М., 1975; Комаров Ф. И., Коровкин Б. Ф. и Меньшиков В. В. Биохимические исследования в клинике, Л., 1976; Липиды, под ред. С. Е. Северина, М., 1977; Меркулов Г. А. Курс патологогистологической техники, с. 241, Л., 1969; П и р с Э. Гистохимия, пер., с англ., с, 259, М., 19 62; Lipids, ed. by R. Paoletti а. о., v. 1—2, N. Y., 1976; Masoro E. J. Physiological chemistry of lipids in mammals, Philadelphia, 1968; Searcy R. L. Lipopa-thies, Springfield, 1971.
Биосинтез холестерина
Ферментативный процесс образования ХС – достаточно сложная «многоходовая комбинация», насчитывающая более 35 энзиматических реакций. Очевидно, что охватить подобный объем преобразований не под силу даже Остапу Бендеру. Поэтому проще рассмотреть базовые стадии биосинтеза холестерола:
- Получение мевалоновой кислоты. Происходит в эукариоте – домене живых организмов. Требует три молекулы активного ацетата.
- Формирование сквалена. Прекурсором выступает ранее произведенная мевалоновая кислота. Изначально соединение трансформируется в активный изопреноид, из 6 молекул которого и образуется сквален.
- Синтез холестерина. Процесс осуществляется циклизацией сквалена. Синтезируется своеобразный прекурсор – ланостерин, переход которого в ХС все еще находится под изучением.
Первоначально биосинтез инициируется формированием ацетоацетил-КоА. Далее, структура подвергается конденсации с 3-ей молекулой активного ацетата. Полученное производное вещество вступает в реакцию восстановления, что и приводит к формированию мевалоната.
Следующий шаг в биосинтезе ХС – превращение мевалоната в сквален. Изначально, соединение подвергается переносу остатка фосфорной кислоты при помощи фермента АТФ. Продуктом реакции выступает 5′-пирофосфорный эфир. Впрочем, соединение не долговечно. Оно практически моментально трансформируется, в эфир мевалоната. Цепочка последующих преобразований достаточно запутана, поэтому проще ограничиться фактом. Результатом этих процессов становится образование сквалена. Реакция циклизации соединения приводит к формированию ланостерина, с последующим биосинтезом холестерола.
Остается добавить, что преимущественно процесс протекает в печени. Поэтому дисфункции органа, способны вызывать нарушения липидного баланса. При нормальной работе, печень производит ежесуточную норму холестерина, с учетом вещества, поступающего с продуктами. Этот факт еще раз опровергает распространенное заблуждение о вреде пищи с высоким содержанием холестерина. Здоровая печень, способна самостоятельно контролировать уровень вещества.
https://youtube.com/watch?v=mzIeQo26NWI
Регуляция плазменного содержания апо-B-липопротеина в норме и при нарушениях липидного обмена
Концентрация апо-B-содержащих липопротеидов в плазме крови является результатом метаболического регулирования их продукции, изменения клиренса или конверсии. Наиболее типичные виды метаболического регулирования содержания апо-В представлены на рис. 2.4. В физиологических условиях, ЛПОНП1 являются основными липопротеидами, секретирующимися гепатоцитами. Непосредственно после синтеза ЛПОНП1 конвертируются в ЛПОНП2, а последние — в ЛППП, при этом их концентрация в плазме крови сохраняется достаточно низкой именно за счет высокой скорости конвертации. С другой стороны, ЛПНП значительно медленнее выводятся из циркуляции (период циркуляции составляет 2–4 сут), что приводит к относительному преобладанию этого типа липопротеидов по отношению к другим фракциям. С возрастом продукция апо-В-липопротеина обычно повышается, что ассоциируется со снижением экспрессии рецепторов к ЛПНП (Matthan N.R. et al., 2005). При некоторых состояниях, например, при ожирении, синтез ЛПОНП особенно высок, что способствует увеличению плазменного уровня апо-В и ЛПНП (Chan D.C. et al., 2004). Установлено, что повышение уровня ЛПОНП1 может сопровождаться гиперинсулинемией даже у здоровых лиц (Malmstrom R. et al., 1997). При инсулинорезистентности недостаточная регуляция продукции ЛПОНП негативно отражается на плазменном уровне апо-B-липопротеина, что способствует повышению концентрации ЛПОНП, ЛППП, ЛПНП, формируя проатерогенную гиперлипидемию (Taskinen M.R., 2003).
Рис. 2.4. Метаболическая регуляция уровня апо-B-липопротеина в плазме крови. Модифицирован из работы C.J. Packard и соавт., 2000
У лиц со значительным повышением уровня ХС ЛПНП (>4,5 ммоль/л) в плазме крови выявляют увеличение содержания апo-B-липопротеинов преимущественно за счет редукции клиренса рецепторов к ним. Последние представляют собой регулируемые мембранассоциированные протеины, ответственные за распознавание и интернационализацию липопротеинов, а также активное поступление ХС в клетки (Turley S.D., 2004). Снижение экспрессии рецепторов или различные варианты качественного изменения в структуре их субъединиц являются морфологической основой возникновения ряда наследственных нарушений липидного обмена, таких как семейная гиперхолестеринемия (см. главу 6). Необходимо отметить, что концентрация ЛПНП может подвергаться непосредственному метаболическому регулированию со стороны ЛПОНП2, уровень которых, в свою очередь, зависит от ЛПОНП1 (Gaffney D. et al., 2002).
У пациентов с изолированной гипертриглицеридемией обычно отмечают избыточную продукцию ЛПОНП (особенно ЛПОНП1) на фоне неэффективного липолиза (Chan D.C. et al., 2004). Это приводит к повышению содержания апо-В в плазме крови, поскольку последний является транспортной формой как для ЛПОНП1, так и для ЛПОНП2. Кроме того, у таких больных не только существенно снижен клиренс апо-В-содержащих липопротеидов, но и имеется возможность для их более интенсивной модификации. Так, CETP транспортирует ТГ от ЛПОНП к ЛПНП и ЛПВП, тогда как эстерифицированный ХС перемещается в обратном направлении. При этом апо-C-II, апо-C-III и апо-Е поочередно включаются в состав липопротеидов, заменяя друг друга. Все это приводит к тому, что в длительно циркулирующих в плазме крови молекулах ЛПОНП снижается содержание aпo-C-липопротеина, тогда как удельный вес aпo-E и эстерифицированного ХС возрастает. Подобная модификация ЛПОНП способствует появлению резистентности последних к липазе и обусловливает формирование так называемых ремнантных форм ЛПОНП (Havel R.J., 2000).
Обратный транспорт ХС
Обратный транспорт ХС — позитивный процесс, с помощью которого он возвращается из периферических тканей в печень для дальнейшего катаболизма (см. рис. 2.2). По современным представлениям, незрелые частицы ЛПВП — хорошие акцепторы свободного ХС. Свободный ХС на поверхности ЛПВП эстерифицируется с образованием эфиров ХС. В роли катализатора эстерификации свободного ХС выступает фермент лецитин-холестерин-ацетилтрансфераза (ЛХАТ), а в качестве кофактора — апопротеин А1, структурный белок ЛПВП (Matsuura F. et al., 2006). Образованные эфиры ХС перемещаются с поверхности частиц ЛПВП в гидрофобное ядро, освобождая, таким образом, дополнительную поверхность для свободного ХС (Tall A.R., 2008). По мере накопления в ядре эфиров ХС, дисковидные частицы ЛПВП преобразуются в сферические, богатые ХС ЛПВП. Эфиры ХС из ЛПВП и содержащих апопротеин В липопротеидов захватываются гепатоцитами через рецептор-опосредованный эндоцитоз или с помощью скавенджер-рецепторов макрофагов/фагоцитов (O’Connell B.J. et al., 2004). Кроме того, эфиры ХС транспортируются от ЛПВП к липопротеидам других классов с помощью специфического белка-переносчика — протеина, транспортирующего эстерифицированный ХС (cholesteryl ester transfer protein — CETP). Мутация гена CETP, приводящая к потере физиологической функции последнего, способствует аккумуляции эфиров ХС в ЛПВП с повышением концентрации последних (Shimoji E. et al., 2004). Кроме того, в молекуле ЛПВП возрастает содержание апо-А1-липопротеина, неэстерифицированного ХС и фосфолипидов. В результате возникает метаболическое ремоделирование ЛПВП, инициирующее интенсивность транспорта ХС с помощью апо-В100-содержащих липопротеидов, в том числе ЛПНП. Это отражает существование тесной взаимосвязи между апо-Е/ABCA1- и CETP-зависимыми процессами транспорта липидов в составе молекул липопротеидов различной плотности (рис. 2.3).
Рис. 2.3. Взаимосвязь между апо-Е-/ABCA1- и CETP-зависимыми процессами транспорта липидов в составе молекул липопротеидов различной плотности
Таким образом, основной детерминантой, обеспечивающей интенсивность транспорта, деградации и модификации липидов в составе липопротеидов, является изменение плотности, формы и архитектоники последних в зависимости от характера метаболического регулирования. В свою очередь, этот процесс тесно ассоциирован с особенностями генотипа, фенотипа, пола, возраста и метаболическими потребностями организма в целом.
Эмульгирование и гидролиз липидов
Первые два этапа переваривания липидов, эмульгирование и гидролиз, происходят практически одновременно. Вместе с этим, продукты гидролиза не удаляются, а оставаясь в составе липидных капелек, облегчают дальнейшее эмульгирование и работу ферментов.
Переваривание в ротовой полости
У взрослых в ротовой полости переваривание липидов не идет, хотя длительное пережевывание пищи способствует частичному эмульгированию жиров.
Переваривание в желудке
Собственная липаза желудка у взрослого не играет существенной роли в переваривании липидов из-за ее небольшого количества и того, что ее оптимум рН 4,5-5,5. Также влияет отсутствие эмульгированных жиров в обычной пище (кроме молока).
Тем не менее, у взрослых теплая среда и перистальтика желудка вызывает некоторое эмульгирование жиров
При этом даже низко активная липаза расщепляет незначительные количества жира, что важно для дальнейшего переваривания жиров в кишечнике, т.к. наличие хотя бы минимального количества свободных жирных кислот облегчает эмульгирование жиров в двенадцатиперстной кишке и стимулирует секрецию панкреатической липазы
Переваривание в кишечнике
Под влиянием перистальтики ЖКТ и составных компонентов желчи пищевой жир эмульгируется. Образующиеся при переваривании лизофосфолипиды также являются хорошим поверхностно-активным веществом, поэтому они способствуют дальнейшему эмульгированию пищевых жиров и образованию мицелл. Размер капель такой жировой эмульсии не превышает 0,5 мкм.
Гидролиз эфиров ХС осуществляет холестерол-эстераза панкреатического сока.
Роль колипазы в действии липазы |
Переваривание ТАГ в кишечнике осуществляется под воздействием панкреатической липазы с оптимумом рН 8,0-9,0. В кишечник она поступает в виде пролипазы, для проявления ее активности требуется колипаза, которая помогает липазе расположиться на поверхности липидной капли.
Колипаза, в свою очередь, активируется и затем образует с липазой комплекс в соотношении 1:1. Панкреатическая липаза отщепляет жирные кислоты, связанные с С1 и С3 атомами углерода глицерола. В результате ее работы остаются 2-моноацилглицеролы (2-МАГ), которые всасываются или превращаются моноглицерол-изомеразой в 1-МАГ. Последний гидролизуется до глицерола и жирной кислоты. Примерно 3/4 ТАГ после гидролиза остаются в форме 2-МАГ и только 1/4 часть ТАГ гидролизуется полностью.
Полный ферментативный гидролиз триацилглицерола
В панкреатическом соке также имеется активируемая фосфолипаза А2, отщепляющая в фосфолипидах жирную кислоту от С2, также обнаружена активность фосфолипазы С и лизофосфолипазы.
Действие фосфолипазы А2 и лизофосфолипазы на примере фосфатидилхолина
В кишечном соке также имеется активность фосфолипазы А2 и фосфолипазы С.
Для работы всех указанных гидролитических ферментов в кишечнике необходимы ионы Са2+, способствующие удалению жирных кислот из зоны катализа.
Точки действия фосфолипаз
Биохимические методы исследования
Биохим, определение Л. проводится гл. обр. в плазме или сыворотке крови, значительно реже в кале (с целью диагностики стеатореи) и моче (при липурии). Определение Л
в плазме крови особенно важно при заболеваниях, сопровождающихся повышением их концентрации в крови (гиперлипидемиях). К ним относятся некоторые заболевания печени (острые и хрон, гепатиты, цирроз и др.), липоидный нефроз (нефротическая гиперлипидемия), сахарный диабет, атеросклероз, панкреатиты, гипотиреоз
Широко применяется определение Л. (холестерина и триглицеридов) в крови при фенотипировании первичных и вторичных гиперлипопротеинемий с целью диагностики и рационального диетического и медикаментозного лечения. Снижение содержания Л. в крови (гиполипидемия) наблюдается реже — при длительном голодании или резко ограниченном потреблении жиров и при гипертиреозе.
При исследовании Л. в крови необходимо строго придерживаться следующих общих принципов: 1) взятие крови производится натощак спустя 10—12 час. после последнего приема пищи; 2) плазма (сыворотка) крови, используемая для анализа, не должна быть гемолизированной; 3) для экстрагирования Л. применяются органические растворители высокой степени очистки; 4) стандарты или референтные препараты Л. сопоставляют с международными стандартами и хранят в замороженном состоянии.
Существует несколько методов определения общих Л. в плазме (сыворотке) крови. Широкое применение нашли гравиметрические методы, основанные на экстрагировании Л. из плазмы крови смесью органических растворителей, с последующим их выпариванием и взвешиванием липидного остатка. Эти методы, однако, не отличаются высокой точностью.
Ряд методов основан на окислении общих Л. хромовой кислотой с последующим титриметрическим или колориметрическим количественным определением (см. Колориметрия, Титриметрический анализ). Широко применяется метод, основанный на цветной реакции, к-рую дают продукты распада Л. с сульфофосфованилиновым реактивом. Метод определения общих Л. в сыворотке крови с сульфофосфованилиновым реактивом принят у нас в стране в качестве унифицированного; содержание Л. в сыворотке крови здорового человека, определенное этим методом, в среднем составляет 350—800 мг%.
Концентрацию общих Л. в сыворотке крови определяют также методом Свана в модификации Л. К. Баумана (окрашенные судаковым черным Л. количественно извлекаются из сыворотки крови и определяются фотометрически) и турбидиметрическим методом (метод Хуэрго), в основу к-рого положено измерение оптической плотности жировой эмульсии, образуемой при взаимодействии серной к-ты с n-диоксановым экстрактом Л. сыворотки крови. Методом Хуэрго в сыворотке крови здорового человека определяется 500 — 700 мг% общих Л.
Для определения триглицеридов наиболее часто применяют методы, в основе которых лежит гидролитическое расщепление триглицеридов. Образовавшийся в результате гидролиза глицерин окисляют до формальдегида и последний определяют колориметрически. Наибольшей точностью из таких методов обладает метод Карлсона, часто применяемый в модификации Игнатовской (H. Ignatowsca).
Для определения холестерина используют методы, основанные на цветной реакции Либерманна— Бурхарда (см. Либерманна-Бурхарда реакция), причем наибольшей точностью из них обладает метод Абелля (см. Абелля метод). Кроме того, для определения холестерина и триглицеридов в крови начинают применять высокоспецифические энзиматические методы с использованием готовых наборов реактивов. Наконец, для определения этих Л. используют автоанализаторы — отечественный прибор АБМ-1, автоанализатор АА-2 фирмы «Техникой» и др. (см. Автоанализаторы).
Методы определения фосфолипидов основаны на экстрагировании или осаждении фосфолипидов из плазмы (сыворотки) крови, минерализации фосфолипидного фосфора, проведении цветной реакции на фосфор и колориметрическом измерении интенсивности окраски (см. Блура метод).
Для определения неэтерифицированных жирных к-т используют титриметрические и колориметрические методы. Из последних наиболее часто применяют методы, основанные на том, что жирные к-ты образуют с медью соли, которые в свою очередь образуют цветные комплексы с диэтил дитиокарбаматом натрия и другими соединениями.
Для разделения Л. используют методы тонкослойной хроматографии, часто с последующим анализом жирных к-т с помощью газожидкостной хроматографии (см. Хроматография).