Мышечная дистрофия дюшенна
Содержание:
- Психотерапия
- Диагностика мышечной дистрофии
- Описание заболевания
- Благотворительные фонды помогают приехать на лечение в Израиль
- Патологическая анатомия
- Лекарства при миодистрофии Дюшенна
- Диагностика Прогрессирующей мышечной дистрофии Дюшенна:
- Какое лечение?
- Причина заболевания
- Мышечная дистрофия
- Признаки патологии
- Питание при атрофии мышц
- Online-консультации врачей
- Диагностика
- Симптомы Прогрессирующей мышечной дистрофии Дюшенна:
- Патогенез
- Лечение дистрофии Дюшена в Германии
- Ваня выздоровел
Психотерапия
Начало клинических проявлений и последующие ухудшения, как правило, провоцируются психическим стрессом. Более того, тяжелое заболевание мышц, само по себе, серьезный психический стресс. На фоне болезни часто развиваются вторичные депрессии с апатией и нежеланием заниматься своим здоровьем. А это ведет к прогрессированию болезни и возможной гибели пациента.
Если это нужно, мы проводим нашим пациентам курс психотерапевтического лечения. Наши пациенты становятся более устойчивыми к психическим раздражителям, становятся активнее, позитивно относятся к лечению, и это незамедлительно сказывается на работе мышц.
Кроме того, мы обучаем пациентов специальным психотехникам для работы с собственными мышцами.
При наличии признаков депрессии возможно назначение современных антидепрессантов (селективные ингибиторы обратного захвата серотонина, такие как Ципралекс, Флуоксетин и др.). Эти антидепрессанты не снижают, а повышают активность пациента и настроение, не вызывают мышечной слабости.
Диагностика мышечной дистрофии
- Аппаратные исследования: МРТ мышечной ткани применяется для определения степени поражений, ЭНМГ (электронейромиография) позволяет оценить функциональные характеристики периферической НС и мышц.
- Лабораторная диагностика: проводится биохимический АК на КФК (уровень креатинфосфокиназы) – увеличенная до 50 раз активность КФК при миодистрофических поражениях свидетельствует о прогрессировании заболевания.
Но наиболее показательными и информативными являются генетические молекулярные исследования (панель «Нервно-мышечные заболевания», микроматричный анализ – ХМА), которые точно определяют нозологическую форму мышечной дистрофии.
Кроме того, около трети диагностируемых случаев миодистрофии Дюшенна имеют спорадический характер. Возникновение 6-7% спонтанных мутаций обусловлено органным гонадным мозаицизмом – наличием у матери генетически здоровых и мутантных первичных половых клеток.
Поэтому пренатальная молекулярная диагностика позволяет идентифицировать, какой именно структурный вариант гена (аллель) получил ребенок. Даже если установлен мужской пол эмбриона, вероятность передачи от матери мутантной популяции гамет не оценивается в 100%, как и генетический риск наследования идентичной мутации братьями (сестрами) больного со спорадической миодистрофией.
Описание заболевания
 Паралич Дюшена-Эрба представляет собой дегенерацию нервного сплетения в области плеча. При развитии аномалии наблюдается нарушение тонуса мускулатуры, потеря чувствительности и атрофия мускулатуры данной области. Самой распространенной является вторично-врожденная форма заболевания, развивающаяся после травмирования ребенка во время родовспоможения. Клиническими проявлениями выступают скованность при попытках отвести, поднять, согнуть в локте верхнюю конечность. Чувствительность и двигательная функциональность в пальцах при этом сохраняется.
Паралич Дюшена-Эрба представляет собой дегенерацию нервного сплетения в области плеча. При развитии аномалии наблюдается нарушение тонуса мускулатуры, потеря чувствительности и атрофия мускулатуры данной области. Самой распространенной является вторично-врожденная форма заболевания, развивающаяся после травмирования ребенка во время родовспоможения. Клиническими проявлениями выступают скованность при попытках отвести, поднять, согнуть в локте верхнюю конечность. Чувствительность и двигательная функциональность в пальцах при этом сохраняется.
Термин и определение патологии были введены и описаны европейскими медиками в начале 70-х годов 19 века. По их именам заболевание и было названо. Оба специалиста выявили прямую связь нарушений двигательной функции в верхнеплечевом сплетении у младенцев с проведенными вспомогательными родами. В современной медицинской литературе встречается и такое определение, как «проксимальный верхний паралич». По статистическим данным нарушение имеет встречаемость в 1-2 случая на 1000 рожденных детей. Разработкой методов предотвращения травмирования младенцев занимается целый ряд специалистов из различных областей медицины: неврологи, ортопеды, травматологи, педиатры, неонатологи, гинекологи и акушеры.
Благотворительные фонды помогают приехать на лечение в Израиль
Международный отдел медицинского центра Хадасса работает в тесном контакте с ведущими благотворительными фондами, которые уже обеспечили десятки больных редкими заболеваниями детей средствами, необходимыми для прохождения лечения в МЦ Хадасса. Мы настоятельно советуем всем родителям, которые взвешивают возможность привести своих детей на лечение в Израиль, обратиться в эти организации.
- Русфонд
- World Vita
- Благотворительный фонд «Алеша»
- Благотворительный фонд «Артемка»
- Благотворительный фонд «Клуб добряков»
- Благотворительный фонд «Помогать легко»
У вас возникли вопросы? Обращайтесь к нам и получите ответ
по телефону: +972 2 560-97-99 (круглосуточно)
по электронной почте: ru-office@hadassah.org.il
или заполнив контактную форму
Патологическая анатомия
Морфологические изменения при Миопатии характеризуются нарастающей атрофией скелетных мышц, к-рые уменьшаются в объеме и становятся плотными, бурого цвета вследствие разрастания соединительной ткани или, напротив, увеличиваются в объеме за счет жировой клетчатки.
Рис. 1. Микропрепарат мышцы (поперечный срез) при миопатии Дюшенна в стадии частичной сохранности двигательной функции: беспорядочное расположение разнокалиберных мышечных волокон — атрофированных (1), нормального диаметра (2), единичных гипертрофированных (3); дистрофические изменения в части мышечных волокон (4); разрастание соединительной ткани в эндомизии (5); гематоксилин-эозин; X 200.
Рис. 2. Микропрепарат мышцы (продольный срез) при миопатии Дюшенна в стадии обездвиженности: единичные атрофированные мышечные волокна (1) среди фиброзной и жировой ткани; лимфоидно-гистиоцитарная инфильтрация (2); гематоксилин-эозин; X 200.
При различных формах мышечных дистрофий (псевдогипертрофических, плече-лопаточно-лицевой, тазоплечевой, офтальмоплегической, бульбарно-офтальмоплегической) определяются в основном однотипные гистол, изменения (рис. 1): уменьшение количества мышечных волокон в пучках, резкая диффузная разнокалиберность сохранившихся волокон с преобладанием среди них атрофированных, гиалиновая и вакуольная дистрофия в части мышечных волокон, дискоидный и коагуляционный некроз отдельных волокон, расщепление гипертрофированных волокон, разрастание соединительной и жировой ткани в эндо- и перимизии. В нек-рых мышечных волокнах находят саркоплазматические тельца, саркоплазматические массы, кольцевидные миофибриллы. Изредка встречаются регенерирующие волокна с круглыми сочными ядрами и богатой рибонуклеопротеидами саркоплазмой. Мышечные веретена длительное время остаются неизмененными. В части наблюдений выявляются периваскулярные лимфоидно-гистиоцитарные инфильтраты. По мере нарастания двигательных нарушений отмечается постепенное уменьшение количества мышечных волокон и нивелировка их диаметра за счет резкого уменьшения количества и калибра гипертрофированных волокон. Наиболее быстро эти изменения развиваются при миопатии Дюшенна — злокачественном варианте псевдогипертрофической М. Для доброкачественного варианта — миопатии Беккера — характерно сочетание выраженных процессов липоматоза н склероза с резкой гипертрофией части мышечных волокон. С последней особенностью связывают длительную компенсацию двигательных нарушений у таких больных. В далеко зашедшей стадии М. определяются небольшие островки из атрофированных мышечных волокон на фоне резкого склероза и липоматоза эндо- и перимизия (рис. 2), при этом гистологически не представляется возможным дифференцировать М. с нейромышечной атрофией. У пробандов при биопсии мышц находят единичные атрофированные мышечные волокна преимущественно I типа, пролиферацию ядер с переходом их в центр волокна и незначительное увеличение соединительной ткани в эндомизии.
Наиболее ранние ультраструктурные изменения в мышцах при Миопатии характеризуются утолщением и расщеплением Z-линий с последующим разрушением миофибрилл мышечного волокна. Благодаря фазово-контрастной и электронной микроскопии выявлены дефекты в мембранных системах мышечного волокна. Гистоферменто химически отмечается ослабление реципрокных отношений между гликолитическими и окислительными ферментами в мышечных волокнах различного типа.
В центральной и периферической нервной системе изменений не обнаружено. Характерен кардиосклероз.
Лекарства при миодистрофии Дюшенна
Медикаментозное лечение является важной частью общей терапии. Наиболее популярной группой препаратов для пациентов с миодистрофией Дюшенна являются стероиды: преднизолон, дефлазакорт и другие
Начинают их прием еще в возрасте 4-6 лет, когда проявляются признаки болезни, но существенного ухудшения состояния нет. Стероиды помогают отсрочить развитие тяжелых симптомов, но в долгосрочной перспективе они неэффективны. Как только у пациента начинает прогрессировать атрофия, улучшить состояние мышц эти лекарства уже не могут. И все же стероиды остаются важной составляющей лечения дистрофии Дюшенна.
Существуют и другие разработки, направленные на улучшение работы мышц. Так, в 2016 году FDA (Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США) одобрило лекарство Exondys51 (этиплирсен), которое сможет стать конкурентом стероидам. По данным проведенных исследований, вещество способно укрепить мышечную ткань, ведь оно увеличивает уровень дистрофина в скелетных мышцах. Однако не все врачи разделяют мнение FDA, поскольку утверждение препарата проходило по ускоренной процедуре. По факту сейчас нет однозначных доказательств, что он может отсрочить развитие паралича или ослабить симптомы развитого заболевания. А значит, Exondys51, как и стероиды, проявляет эффективность лишь в коротком периоде болезни. Несмотря на то, что лекарство уже утверждено, FDA продолжает исследования. Если эффективность препарата не будет подтверждена, его производство остановят.
Европейский Комитет по лекарственным препаратам для человека (CHMP) в 2014 году зарегистрировал другой медикамент — Трансларна (аталурен). В отличие от других лекарств, которые только укрепляют мышцы, аталурен способен восстанавливать нарушенный синтез белков. А значит, теоретически устраняет саму причину развития болезни. Поскольку это новая разработка, понять, насколько она эффективна в долгосрочной перспективе, пока трудно. Но промежуточные результаты достаточно обнадеживающие.
Диагностика Прогрессирующей мышечной дистрофии Дюшенна:
Диагноз ставится на основании данных генеалогического анализа (рецессивный сцепленный с Х-хромосомой тип наследования), клинических обострений болезни (раннее начало в 1–3 года, симметричные атрофии проксимальных групп мышц, развивающиеся в восходящем направлении, псевдогипертрофии икроножных мышц, грубые соматические и нейроэндокринные расстройства, снижение интеллекта, быстрое злокачественное течение болезни), данных биохимических исследований (типично раннее, с 5-го дня жизни ребенка, увеличение активности КФК – в 30–50 раз выше нормы), игольчатой электромиографии и морфологических результатов. позволяющих выявить первично-мышечный (миодистрофический) тип поражения.
Дифференцировать заболевание следует от спинальной амиотрофии Верднига– Гоффманна, рахита, врожденного вывиха бедра.
Какое лечение?
Единственным доступным видом фармакологического лечения
является терапия кортикостероидами (преднизолон или дефлазакорт). Гормоны
улучшают мышечную функцию, помогают предупредить осложнения .
Точный механизм действия кортикостероидов при дистрофии Дюшенна
до сих пор неизвестен, но предполагается, что они блокируют разрушение
(протеолиз) мышечных волокон, стимулируют восстановление мышечной ткани (стимулируют
активность миофибробластов, стабилизируют мембрану мышечных волокон), оказывают
противовоспалительное и иммуносупрессивное действие (подавляют клеточные и
гуморальные иммунные реакции, которые возникают в разрушенных мышечных волокнах
из-за отсутствия дистрофина, действия оксидативного стресса и пр.) .
Стероиды рекомендуется назначать после окончательного
формирования двигательных навыков, но до начала их угасания, т.е. нарушения
двигательных функций, связанных с заболеванием. Обычно кортикостероиды
назначаются в возрасте 4—6 лет .
До старта лечения препаратами желательно провести вакцинацию
в соответствии с национальным календарем. Вакцинация живыми вакцинами на фоне
длительного приема стероидов не проводится .
Врачу важно проинформировать семью о возможных побочных
эффектах терапии и тщательно проводить мониторинг за состоянием ребенка. Кроме
того, необходимо предупредить семью о недопустимости самостоятельного отказа от
приема препаратов или пропуске очередной дозы (это чревато развитием острой
надпочечниковой недостаточности — нечастым, но грозным осложнением)
Использование анаболических стероидов, ботулотоксина,
креатина и различных биологических добавок (коэнзим Q, карнитин, аминокислоты, антиоксиданты
и пр.) не рекомендовано для лечения пациентов с мышечной дистрофией Дюшенна.
Эффективность указанных препаратов не исследована и расценивается как
сомнительная .
Большое внимание в лечении детей необходимо уделять
физической реабилитации и психосоциальной помощи. Для профилактики мышечной
атрофии настоятельно рекомендуется плавание и/или езда на велосипеде
Появление
выраженных мышечных болей и/или миоглобинурии (появление окрашенной мочи) после
занятий спортом говорит о чрезмерном уровне физической активности .
Причина заболевания
В основе мышечной дистрофии Дюшенна лежит генетический дефект половой Х хромосомы.
Один из участков Х хромосомы содержит ген, кодирующий производство в организме особого мышечного белка под названием дистрофин. Белок дистрофин составляет основу мышечных волокон (миофибрилл) на микроскопическом уровне. Функция дистрофина заключается в поддержании клеточного скелета, в обеспечении способности миофибрилл к многократным актам сокращения и расслабления. При мышечной дистрофии Дюшенна этот белок либо отсутствует вообще, либо синтезируется дефектным. Уровень нормального дистрофина не превышает 3%. Это приводит к разрушению мышечных волокон. Мышцы перерождаются и заменяются жировой и соединительной тканью. Естественно, что при этом утрачивается двигательный компонент человеческой деятельности.
Заболевание наследуется по рецессивному типу, сцепленному с Х хромосомой. Что это означает? Поскольку все гены человека парные, то есть дублируют друг друга, то для того, чтобы появились патологические изменения в организме при наследственном заболевании, необходимо, чтобы генетический дефект возник в одной хромосоме или аналогичных участках обеих хромосом. Если заболевание возникает только при мутациях в обеих хромосомах, то такой тип наследования называют рецессивным. Когда же генетическая аномалия выявляется только в одной хромосоме, но болезнь все равно развивается, такой тип наследования называют доминантным. Рецессивный тип возможен только при одновременном поражении идентичных хромосом. Если вторая хромосома будет «здорова», то заболевание не возникнет. Именно поэтому мышечная дистрофия Дюшенна является уделом лиц мужского пола, потому что они имеют в генетическом наборе одну Х хромосому, а вторую (парную) – У. Если мальчику попадается «поломанная» Х хромосома, то у него обязательно возникает болезнь, потому что здоровой хромосомы у него просто нет. Для того, чтобы мышечная дистрофия Дюшенна возникла у девочки, необходимо совпадение в ее генотипе двух патологических Х хромосом, что практически маловероятно (в таком случае папа девочки должен быть болен, а у мамы в генетическом наборе должна содержаться дефектная Х хромосома). Девочки выступают лишь носителями заболевания и передают его своим сыновьям. Конечно, часть случаев заболевания не является результатом передачи по наследству, а возникает спорадически. Это означает появление мутации в генетическом наборе ребенка спонтанно. Вновь появившаяся мутация может быть передана по наследству (при условии сохранения способности к размножению).
Мышечная дистрофия
Мышечная дистрофия, группа хронических наследственных заболеваний скелетных (произвольных) мышц человека. Мышечные дистрофии проявляются прогрессирующей слабостью и дегенерацией мышц.
Существует целый ряд форм мышечной дистрофии. Они различаются по таким характеристикам, как возраст, в котором начинается заболевание, локализация пораженных мышц, выраженность мышечной слабости, скорость прогрессирования дистрофии и тип ее наследования. Чаще всего встречаются две формы: мышечная дистрофия Дюшенна и миотоническая мышечная дистрофия.
Мышечная дистрофия Дюшенна
(псевдогипертрофическая мышечная дистрофия) – самая частая форма этого заболевания у детей. Причина болезни – генетический дефект, локализованный на X-хромосоме (одной из двух хромосом, определяющих пол человека).
Женщины с дефектным геном передают его своим детям, но у них самих симптомы дистрофии, как правило, отсутствуют. У мальчиков, получивших дефектный ген, в возрасте от двух до пяти лет неизбежно развивается мышечная слабость.
В первую очередь страдают крупные мышцы нижних конечностей и тазового пояса. Затем дегенерация распространяется на мышцы верхней половины тела, а далее постепенно и на все основные группы мышц.
Характерное проявление болезни – псевдогипертрофия икроножных мышц, т.е. увеличение их за счет отложения жира и разрастания соединительной ткани.
Мышечная дистрофия Дюшенна – одна из самых тяжелых и быстро прогрессирующих форм. К 12 годам больные обычно теряют способность передвигаться, а к 20 годам большинство из них погибает.
Миотоническая мышечная дистрофия
(болезнь Штейнерта) – наиболее распространенная форма мышечной дистрофии у взрослых. Она обусловлена дефектным геном на 19-й хромосоме. Мужчины и женщины страдают в равной степени и могут передать генетический дефект детям.
Заболевание проявляется в любом возрасте, в том числе в младенчестве, но чаще всего – между 20 и 40 годами. Первыми симптомами служат миотония (замедленное расслабление мышц после сокращения) и слабость мимических мышц; возможно также поражение мышц конечностей и других частей тела.
Прогрессирование болезни происходит в большинстве случаев медленно, и полная инвалидность может наступить не ранее чем через 15 лет.
Особенность данного заболевания состоит в том, что помимо произвольных мышц оно поражает также гладкую мускулатуру и сердечную мышцу.
Патоморфология
Все формы мышечной дистрофии характеризуются дегенерацией мышц, но не связанных с ними нервов. В пораженной мышечной ткани обнаруживают различные изменения, в том числе значительные колебания толщины (диаметра) мышечных волокон. Постепенно эти волокна теряют способность сокращаться, распадаются и замещаются жировой и соединительной тканью.
Диагноз
По своим клиническим проявлениям мышечные дистрофии сходны со спинальными амиотрофиями – наследственными болезнями, поражающими двигательные нейроны спинного мозга.
Эти болезни тоже приводят к выраженной мышечной слабости, иногда угрожающей жизни.
Для подтверждения диагноза мышечной дистрофии может потребоваться электромиография, а иногда и биопсия мышц с микроскопическим исследованием для выявления характерных дистрофических изменений.
Причины
Специалисты полагают, что каждая форма мышечной дистрофии обусловлена отдельным точечным генетическим дефектом, нарушающим способность мышечных клеток синтезировать необходимые белки.
Усилия исследователей сосредоточены на поиске дефектов, лежащих в основе заболеваний, и тех отклонений в составе белков, к которым эти дефекты приводят.
В настоящее время выявлен ген мышечной дистрофии Дюшенна.
Лечение
Не существует способов предотвратить или замедлить прогрессирование мышечной слабости при мышечной дистрофии.
Терапия направлена главным образом на борьбу с осложнениями, такими, как деформация позвоночника, развивающаяся вследствие слабости мышц спины, или предрасположенность к пневмониям, обусловленная слабостью дыхательных мышц.
В этом направлении достигнуты определенные успехи, и качество жизни больных с мышечной дистрофией улучшилось. Сейчас многие больные, несмотря на свой недуг, могут вести полнокровную и продуктивную жизнь.
Проверь себя!
Ответь на вопросы викторины «Фобии и страхи»
Чего больше всего боятся трискаидекафобы?
Признаки патологии
Клинические проявления болезни подразделяются на три этапа, характеризующиеся разной симптоматикой:
1. Острая стадия. Начинается сразу после травмирования и длиться на протяжении месяца после него. В данный период рука находится в одном положении – разогнута в локте, прижата к телу, пальцы подведены к ладони. Больной не может самостоятельно отвести руку вперед, вверх, в сторону, развернуть внутреннюю поверхность конечности в переднее положение. Любые манипуляции, попытки изменить положение конечности вызывают болезненность и дискомфорт. На пораженной стороне не фиксируется рефлекс бицепса. У младенцев отсутствует хватательный рефлекс на стороне паралича. При помещении ребенка в горизонтальное положение наблюдается «безвольное» свисание руки. Мышечный тонус снижен, кожные покровы холодные и обесцвеченные, сенсорика мало восприимчива к болевым ощущениям, наносимым на кожу.
2. Восстановительная стадия. В зависимости от тяжести травмирования, своевременности терапевтической помощи и адекватности медицинских манипуляций будет проявляться следующая симптоматика. При легком параличе чувствительность и активность постепенно восстанавливается до полной функциональности. Однако у детей к трем годам заметно небольшое укорочение больной руки, слабость мускулатуры.
3. Стадия остаточных явлений. Так как функциональность в восстановительный период возвращается не в полном объеме, развиваются последующие нарушения. Если паралич имеет среднюю или тяжелую форму, больной жалуется на синдром «кукольной руки». Он проявляется четко выраженной складкой между грудиной и плечевой зоной. Меняется структура мышечной ткани, локтевой сустав теряет сгибательную способность. Четко заметен выраженный поворот лопатки, пальца сохраняют ладонное предлежание. Так как мускулатура атрофируется возможен последующий хронический вывих плечевого участка. Следствием измененной анатомии верхнего отдела позвоночного столба становится искривление последнего, сколиозные проявления.
Питание при атрофии мышц
Миопатия и амиотрофия приводят к потере мышечной ткани. Задача пациента и доктора – успевать эту потерю восстанавливать. Известно, что для роста мышц требуется белок, но не все знают какой, в каком количестве и режиме. Мы рекомендуем принимать питательные смеси в виде аминокислот, с добавлением витаминов и L-карнитина. Дозы рассчитываем с учетом веса, возраста пациента и состояния желудочно-кишечного тракта.
Нередко белки по какой-то причине не усваиваются из желудочно-кишечного тракта и прием белкового питания не приводит ни к чему, кроме проблем с пищеварением. Информацию о качестве усвоения белков можно получить в результате копрологического исследования кала. Поскольку питание – важная составляющая лечения миопатии, мы в этих случаях обязательно лечим причину нарушения усвоения белков (снижение экскреторной функции поджелудочной железы, воспаление слизистой кишечника и т.п.).
Online-консультации врачей
| Консультация доктора-УЗИ |
| Консультация детского психолога |
| Консультация семейного доктора |
| Консультация детского невролога |
| Консультация гастроэнтеролога детского |
| Консультация стоматолога |
| Консультация гинеколога |
| Консультация педиатра-аллерголога |
| Консультация репродуктолога (диагностика и лечение бесплодия) |
| Консультация психоневролога |
| Консультация массажиста |
| Консультация нейрохирурга |
| Консультация гомеопата |
| Консультация эндокринолога |
| Консультация уролога |
Новости медицины
Устройство и принцип работы магнитной мешалки,
26.05.2021
Быстрая доставка лекарств на сервисе mednex.com.ua,
30.04.2021
6 простых привычек, чтобы круглый год не болеть простудами: рекомендуют все врачи,
17.03.2021
Морепродукты становятся вредными для здоровья?,
05.01.2021
Новости здравоохранения
Эксперт назвала три отличия простуды от COVID-19,
05.01.2021
В мире более 86 миллионов случаев COVID-19,
05.01.2021
Скорость распространения COVID-19 зависит от климатических условий,
11.06.2020
Исследователи насчитали шесть разновидностей коронавируса,
11.06.2020
Диагностика
Людям с семейным анамнезом заболевания рекомендуется генетическое консультирование. МДД можно обнаружить с точностью около 95% с помощью генетических исследований, проводимых во время беременности. Уровни креатинкиназы (КФК-ММ) в кровотоке чрезвычайно высоки. Электромиографии (ЭМГ) показывает , что слабость , вызванное разрушением мышечной ткани , а не повреждение нервов .
ДНК тест
Специфическая для мышц изоформа гена дистрофина состоит из 79 экзонов , и тестирование ДНК ( анализ крови ) и анализ обычно могут идентифицировать конкретный тип мутации экзона или экзонов, которые затронуты. Анализ ДНК в большинстве случаев подтверждает диагноз.
Биопсия мышц
Если при тестировании ДНК не удается обнаружить мутацию, может быть проведена биопсия мышцы. Небольшой образец мышечной ткани извлекается с помощью иглы для биопсии. Ключевые тесты, выполняемые на образце биопсии на МДД, — это иммуногистохимия , иммуноцитохимия и иммуноблоттинг на дистрофин, и их следует интерпретировать опытным нейромышечным патологом. Эти тесты предоставляют информацию о наличии или отсутствии белка. Отсутствие белка — положительный тест на МДД. Там, где присутствует дистрофин, тесты показывают количество и размер молекулы дистрофина, помогая отличить МДД от более легких фенотипов дистрофинопатии . За последние несколько лет были разработаны тесты ДНК, которые выявляют большее количество мутаций, вызывающих это состояние, и биопсия мышц не требуется так часто, чтобы подтвердить наличие МДД.
Пренатальные тесты
Пренатальный тест может быть рассмотрен, если мать является известным или подозреваемым носителем.
Пренатальные тесты могут определить, есть ли у будущего ребенка одна из самых распространенных мутаций. Многие мутации ответственны за МДД, а некоторые не были идентифицированы, поэтому генетическое тестирование может быть ложноотрицательным, если подозреваемая мутация у матери не идентифицирована.
Перед инвазивным тестированием важно определить пол плода; в то время как мужчины иногда страдают этим Х-сцепленным заболеванием, МДД у женщин встречается крайне редко. Этого можно добиться с помощью УЗИ в 16 недель или позже путем бесплатного анализа ДНК плода
Взятие пробы ворсинок хориона (CVS) можно сделать на 11–14 неделе, и риск выкидыша составляет 1%. Амниоцентез можно провести через 15 недель, при этом риск выкидыша составляет 0,5%. Забор крови плода можно сделать примерно в 18 недель. Другой вариант в случае неясных результатов генетического теста — биопсия мышц плода.
Симптомы Прогрессирующей мышечной дистрофии Дюшенна:
Признаки заболевания проявляются в первые 1–3 года жизни
Уже на 1-м году обращает на себя внимание отставание детей в моторном развитии. Они, как правило, с задержкой начинают садиться, вставать, ходить
Движения неловкие, при ходьбе дети не усто йчивы, часто спотыкаются, падают. В 2–3 года по являются мышечная слабость, патологическая мышечная утомляемость, проявляющаяся при физической нагрузке – длительной ходьбе, подъеме на лестницу, изменение походки по типу «утиной». В этот период обращает на себя внимание своеобразная «стереотипная» динамика движений детей во время вставания из горизонтального положения, из положения на корточках или со стула. Вставание происходит поэтапно, с активным использованием рук – «взбирание лесенкой» или «взбирание по самому себе». Атрофии мышц всегда симметричны. Вначале они локализуются в проксимальных группах мышц нижних конечностей – мышцах тазового пояса, бедер, а через 1– 3 года быстро распространяются в восходящем направлении на проксимальные группы мышц верхних конечностей – плечевой пояс, мышцы спины. Вследствие атрофии появляются лордоз, «крыловидные» лопатки, «осиная» талия. Типичным, «классическим» симптомом заболевания является псевдогипертрофия икроножных мышц.
При пальпации мышцы плотны, безболезненны. У многих больных в результате селективного и неравномерного поражения различных групп мышц рано возникают мышечные контрактуры и сухожильные ретракции. Мышечный тонус снижен преимущественно в проксимальных группах мышц. Сухожильные рефлексы изменяются с различной последовательностью. В ранних стадиях болезни исчезают коленные рефлексы, позднее – рефлексы с двуглавой и трехглавой мышц. Пяточные (ахилловы) рефлексы длительное время остаются сохранными.
Снижение амплитуды осцилляции и увеличение полифазности.
Одной из отличительных особенностей миодистрофии Дюшенна является сочетание данной формы с патологией костно-суставной системы и внутренних органов (сердечно-сосудистой и нейроэндокринной систем). Костно-суставные нарушения характеризуются деформациями позвоночника, стоп, грудины. На рентгенограммах обнаруживаются сужение костно-мозгового канала, истончение коркового слоя длинных диафизов трубчатых костей.
Сердечно-сосудистые расстройства клинически проявляются лабильностью пульса, артериального давления, иногда глухостью тонов и расширением границ сердца. На ЭКГ регистрируются изменения миокарда (блокада нож ек пучка Гиса и др.). Нейроэндокринные нарушения встречаются у 30–50 % больных. Чаще других наблюдаются синдром Иценко– Кушинга, адипозогенитальная дистрофия Бабинского–Фрелиха. Интеллект у многих больных снижен в различной степени.
Течение. Болезнь имеет быстро прогрессирующее злокачественное течение. К 7–10 годам возникают глубокие двигательные расстройства – выраженное изменение походки, снижение мышечной силы, в значительной степени ограничивающие свободное, самостоятельное передвижение больных. К 14–15 годам наступает обездвиженность.
Патогенез
Мышечная миопатия Дюшенна развивается на фоне мутации в гене, кодирующем белок дистрофин в локусе Xp21.2. Практически все дети унаследуют дистрофию от матери-носителя дефектного гена. Причем женщины-носители патологически измененного гена не подвержены миопатии Дюшенна, что обусловлено наличием второй половой хромосомы, обеспечивающей выработку дистрофина. Также генная мутация может возникать в материнской яйцеклетке. Тяжелое течение миопатии Дюшенна обусловлено генетическими аберрациями, приводящими к нарушению считывания дезоксирибонуклеиновой кислоты и абсолютному прекращению синтеза дистрофина, влекущее за собой разрушение мышечных клеток (миоцитов).
Лечение дистрофии Дюшена в Германии
Немецкие специалисты используют консервативные и физиотерапевтические методы лечения миопатии Дюшена. К сожалению, при выявлении этого заболевания на поздних стадиях терапевтический эффект незначителен. Если же этот патологический процесс верифицирован на начальных этапах его развития, то высококвалифицированным специалистам удается приостановить прогрессирование заболевания и значительно улучшить качество жизни пациента.
Лечение миопатии Дюшена должно быть комплексным и влиять на все звенья патогенеза. В клиниках Германии используются следующие фармакологические средства для терапии этого заболевания:
- Глюкокортикостероиды – препараты, которые могут на некоторое время приостановить прогрессирование патологического процесса. Стероиды назначаются непродолжительными курсами с обязательным предупреждением возможных побочных действий.
- Прозерин – препарат, который улучшает передачу нервных импульсов к соответственным участкам мышц. Таким образом мышцы более длительное время поддерживаются в тонусе.
- Витамины также необходимы для улучшения метаболических процессов, поддержки тонуса мышц пациента.
- Препараты кальция. Кальций необходимый для нормального сокращения мышечного волокна. Именно поэтому немецкие специалисты назначают его при миопатии Дюшена.
Также в немецких клиниках используются следующие физиотерапевтические методы лечения миопатии Дюшена:
- ЛФК. Для детей с миопатией Дюшена ограничивают занятия физической культурой. Немецкие специалисты разрабатывают для каждого ребенка индивидуальные курсы массажа и лечебной физкультуры, которые позволяют снять утомление и напряжение с мышц;
- Специальные шины для лодыжек или для голени применяются у пациентов с первичными клиническими проявлениями заболевания. Шины рекомендуется одевать на ночь.
В клиниках Германии пациенты с миопатией Дюшена имеют возможность не только продлить жизнь с помощью эффективных методов лечения, но и предупредить множественные осложнения заболевания. Кроме того, лечащий врач всегда обучает родителей больного ребенка простым манипуляциям по уходу
Для больного ребенка очень важно находиться в комфортных условиях и дружелюбном окружении высококвалифицированных немецких специалистов
Ваня выздоровел
В Украине Ване Волохатюку в течение 2 лет не могли поставить диагноз. В МЦ Хадасса у него обнаружили редкое и очень опасное заболевание – синдром IPEX. Ребенка спасли, проведя успешную трансплантацию костного мозга (ТКМ).
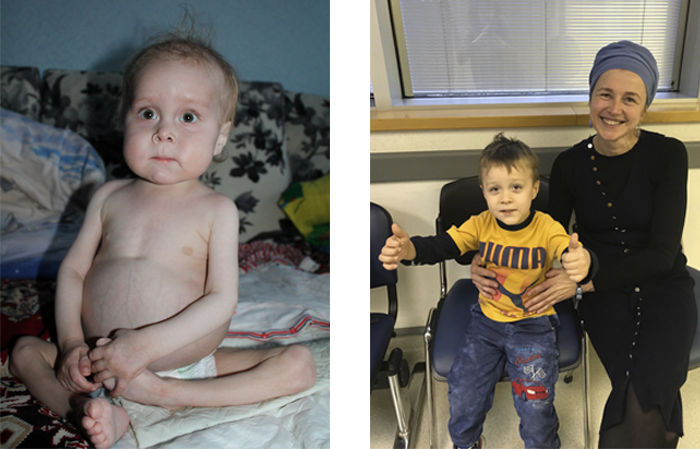
Фото слева: так Ваня выглядел до того, как профессор Полина Степенски успешно провела ему ТКМ в медицинском центре Хадасса.
Фото справа: Ваня после ТКМ с сотрудницей нашего медицинского департамента Эстер Винокуров.
Для справки:
- По разным оценкам в России к 2017 году было зарегистрировано от 1.5 до 5 миллионов больных с редкими заболеваниями.
- В 2017 году в Украине на лечение орфанных заболеваний был выделен 1 млрд. гривен, и эта сумма почти сразу же была признана недостаточной.
- В Беларуси зарегистрированы тысячи случаев орфанных заболеваний, по оценкам местной прессы ими болеют от 6% до 8% населения.