Хронический миелолейкоз
Содержание:
- История [ править ]
- Лечение Острого лимфобластарного лейкоза:
- Что такое Хронический миелоидный лейкоз —
- К каким докторам следует обращаться если у Вас Хронический миелоидный лейкоз:
- Другие заболевания из группы Болезни крови, кроветворных органов и отдельные нарушения, вовлекающие иммунный механизм:
- Что провоцирует / Причины Острого промиелоцитарного лейкоза:
- 1.Что такое хронический миелолейкоз?
- Лейкемоидные реакции: диагностика
- К каким докторам следует обращаться если у Вас Хронический миелолейкоз:
- Лечение хронического лейкоза
- Лечение хронического лимфоцитарного лейкоза (лимфолейкоза) в зависимости от группы риска
- Сопоставимые онкогены
- Фазы хронического миелолейкоза
- Общее описание
- Патогенез хронического миелолейкоза
- Номенклатура [ править ]
- Причины острого миелоидного лейкоза
- К каким докторам следует обращаться если у Вас Сублейкемический миелоз:
История [ править ]
Хромосома Филадельфия была впервые открыта и описана в 1959 году Дэвид Ньюбери в в Институте Ланкенау больницы по исследованию рака , который слился с онкологической больницы американской в 1974 году , чтобы создать Fox Chase Cancer Center , вместе с Питером Nowell из Университета Пенсильвании Школа медицины . Генетическая аномалия, обнаруженная Хангерфордом и Ноуэллом, была названа в честь города, в котором располагались обе организации.
Хангерфорд писал докторскую диссертацию по хромосомам в лаборатории генетики, которая тогда была Институтом исследований рака в Исследовательском институте больницы Ланкенау и обнаружил дефект в хромосомах из клеток крови больных лейкемией. Это основополагающее наблюдение было первым генетическим дефектом, связанным с конкретным раком человека. Ноуэлл был патологом в Пенсильванском университете, который также изучал лейкозные клетки под микроскопом, когда он заметил клетки с этим генетическим дефектом в процессе деления. К его удивлению, их хромосомы — обычно нечеткий клубок — были видны как отдельные структуры. В поисках специалиста по хромосомам Ноуэлл нашел Хангерфорд в Ланкенау. Проводя свои микроскопические исследования, Хангерфорд продолжил свои наблюдения, обнаружив, что некоторые лейкозные клетки имеют аномально короткую хромосому 22. Впоследствии наблюдаемая им мутация стала известна как филадельфийская хромосома.
В 1973 годе Джанет Роули в Университете Чикаго определили механизм , с помощью которого Филадельфийской хромосомы возникающих транслокаций.
Лечение Острого лимфобластарного лейкоза:
Частота улучшений у детей при этой форме лейкоза составляет 94%, у лиц старше 15 лет — около 80%. Частота выздоровления у детей — более 50%. Прогностически неблагоприятными факторами, которые влияют на продолжительность жизни больных острым лимфобластным лейкозом, являются распространенность процесса к моменту постановки диагноза, лейкоцитоз выше 15 Ч 103 в 1 мкл, увеличение селезенки, вовлечение в процесс узлов средостения, раннее поражение центральной нервной системы и возраст моложе 1 года и старше 10 лет.
Проводится немедленное лечение цитостатическими препаратами и только по специальным программам.
Целью лечения острых лейкозов является достижение и максимальное продление улучшения или выздоровление.
Острые лимфобластный и недифференцируемый лейкозы у детей. Лечение проводится по программам (разработанным различными авторами), которые позволяют более чем у 50% детей сохранять улучшение дольше 5 лет.
Улучшение достигается за 4-6 недель с помощью одной из 3 схем, следует отметить, что данные схемы были внедрены еще в 1980-1990 гг. и до сих пор не потеряли свою актуальность.
Винкристин по 1,4 мг/м2 1 раз в 7 дней внутривенно, преднизолон по 40 мг/м2 в день (в схемах, рассчитанных на 4-6 недель, преднизолон отменяют в течение 6-8 дней).
Винкристин по 1,4 мг/м2 1 раз в 7 дней внутривенно, преднизолон по 40 мг/м2 в день, рубомицин по 60 мг/м2 2 дня подряд на 2-й неделе терапии (на 10-й и 11-й дни курса).
Винкристин по 1,4 мг/м2 1 раз в 7 дней внутривенно, преднизолон по 40 мг/м2 в день, L-аспарагиназа в течение 10 дней по 100 ЕД/кг в день внутривенно после 4-6 недель применения винкристина и преднизолона (если нет полного эффекта).
При неэффективности лечения по схеме 1 в течение 4-6 недель (у лиц моложе 10 лет) назначают лечение по схеме 2 или 3.
При отсутствии эффекта от лечения по схемам врач назначает комбинации с онковином или с винбластином.
Закрепляющие курсы проводят 1-3 раза, в зависимости от значительности нарушения условий выполнения лечения в период улучшения, протяженности этого периода, распространенности лейкемического процесса в начале лечения, полноты полученного улучшения. Если врач обнаруживает селезенку в глубине подреберья, то это может послужить основанием для повторения курса закрепляющего лечения. Если селезенка увеличена, то врач ее пунктирует и в случае ее лимфоцитарного состава назначает лечение, направленное на поддержание улучшения.
Сразу после установления диагноза проводят спинномозговую пункцию с введением в спинномозговой канал метотрексата в дозе 12,5 мг/м2; во время улучшения и курса, закрепляющего улучшение, регулярно 1 раз в 2 недели повторяют спинномозговые пункции с введением метотрексата в дозе 12,5 мг/м2. В случае обнаружения любого числа бластных клеток в спинномозговой жидкости начинают лечение нейролейкемии, профилактическое облучение головы отменяется.
Достижение улучшения обязательно подтверждается контрольной пункцией костного мозга; первую после диагностической пункцию костного мозга в период улучшения производят через 7 дней после начала лечения (уменьшение бластоза в этом пунктате на 50% от исходного и более означает хороший прогноз), затем через 4 недели от начала лечения.
Пролиферативная активность лейкозных клеток резко возрастает после периода улучшения, как и после любого цитостатического курса. В связи с этим непосредственно после достижения улучшения врач назначает поддерживающее лечение.
В комбинации в период поддерживания улучшения дозы цитостатических препаратов, исключая винкристин и преднизолон, уменьшают вдвое.
Развитие полиневрита (снижение сухожильных рефлексов, мышечного тонуса, онемение в пальцах рук и ног и в дальнейшем развитие пареза конечностей с атрофией мышц), обусловленное токсическим действием винкристина, требует снижения дозы этого препарата вдвое, а при выраженности или нарастании изменений — замены его винбластином (через несколько недель после отмены препарата полиневрит проходит). Лечение цитостатическими препаратами отменяется при уровне лейкоцитов ниже 1 Ч 103 (1000) в 1 мкл, язвенном стоматите, диарее, тяжелой рвоте, при высокой температуре, сохраняющейся более 2 дней.
Профилактику нейролейкемии при острых лимфобластном и недифференцируемом лейкозах у детей проводят при цитологически нормальном составе спинномозговой жидкости (бластных клеток нет, цитоз менее 10 в 1 мкл) с первой недели улучшения.
Первая схема профилактики: облучение головы в суммарной дозе 24 Гр и параллельно 5 введений метотрексата эндолюмбально. Профилактику можно проводить в основном амбулаторно.
Что такое Хронический миелоидный лейкоз —
Хронический миелоидный лейкоз (ХМЛ) занимает третье место среди всех лейкозов. На его долю приходится около 20% случаев рака крови. На данный момент в России зарегистрировано более 3 тысяч больных. Самому маленькому из них всего 3 года, самому старшему – 90.
Заболеваемость ХМЛ составляет 1-1,5 случая на 100 000 населения в год (15-20% от всех случаев гемобластозов у взрослых). Болеют преимущественно люди среднего возраста: пик заболеваемости приходится на возраст 30-50 лет, около 30% составляют больные старше 60 лет. У детей ХМЛ встречается редко, составляя не более 2-5%о от числа всех лейкозов. Мужчины болеют несколько чаще женщин (соотношение 1:1,5).
К каким докторам следует обращаться если у Вас Хронический миелоидный лейкоз:
Гематолог
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Хронического миелоидного лейкоза, ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору – клиника Eurolab всегда к Вашим услугам! Лучшие врачи осмотрят Вас, изучат внешние признаки и помогут определить болезнь по симптомам, проконсультируют Вас и окажут необходимую помощь и поставят диагноз. Вы также можете вызвать врача на дом. Клиника Eurolab открыта для Вас круглосуточно.
Как обратиться в клинику:
Телефон нашей клиники в Киеве: (+38 044) 206-20-00 (многоканальный). Секретарь клиники подберет Вам удобный день и час визита к врачу. Наши координаты и схема проезда указаны здесь. Посмотрите детальнее о всех услугах клиники на ее персональной странице.
Другие заболевания из группы Болезни крови, кроветворных органов и отдельные нарушения, вовлекающие иммунный механизм:
| B12-дефицитная анемия |
| Анемии, обусловленные нарушением синтеза утилизацией порфиринов |
| Анемии, обусловленные нарушением структуры цепей глобина |
| Анемии, характеризующиеся носительством патологически нестабильных гемоглобинов |
| Анемия Фанкони |
| Анемия, связанная со свинцовым отравлением |
| Апластическая анемия |
| Аутоиммунная гемолитическая анемия |
| Аутоиммунная гемолитическая анемия |
| Аутоиммунная гемолитическая анемия с неполными тепловыми агглютининами |
| Аутоиммунная гемолитическая анемия с полными Холодовыми агглютининами |
| Аутоиммунная гемолитическая анемия с тепловыми гемолизинами |
| Болезни тяжелых цепей |
| болезнь Верльгофа |
| Болезнь Виллебранда |
| болезнь Ди Гулъелъмо |
| болезнь Кристмаса |
| Болезнь Маркиафавы-Микели |
| Болезнь Рандю — Ослера |
| Болезнь тяжелых альфа-цепей |
| Болезнь тяжелых гамма-цепей |
| Болезнь Шенлейн — Геноха |
| Внекостномозговые поражения |
| Волосатоклеточный лейкоз |
| Гемобластозы |
| Гемолитико-уремический синдром |
| Гемолитико-уремический синдром |
| Гемолитическая анемия, связанная с дефицитом витамина Е |
| Гемолитическая анемия, связанная с дефицитом глюкозо-6-фосфатдегидрогеназы (Г-6-ФДГ) |
| Гемолитическая болезнь плода и новорожденного |
| Гемолитические анемии, связанные с механическим повреждением эритроцитов |
| Геморрагическая болезнь новорожденных |
| Гистиоцитоз злокачественный |
| Гистологическая классификация лимфогранулематоза |
| ДВС-синдром |
| Дефицит К-витаминзависимых факторов |
| Дефицит фактора I |
| Дефицит фактора II |
| Дефицит фактора V |
| Дефицит фактора VII |
| Дефицит фактора XI |
| Дефицит фактора XII |
| Дефицит фактора XIII |
| Железодефицитная анемия |
| Закономерности опухолевой прогрессии |
| Иммунные гемолитические анемии |
| Клоповое происхождение гемобластозов |
| Лейкопении и агранулоцитозы |
| Лимфосаркомы |
| Лимфоцитома кожи (болезнь Цезари) |
| Лимфоцитома лимфатического узла |
| Лимфоцитома селезенки |
| Лучевая болезнь |
| Маршевая гемоглобинурия |
| Мастоцитоз (тучноклеточный лейкоз) |
| Мегакариобластный лейкоз |
| Механизм угнетения нормального кроветворения при гемобластозах |
| Механическая желтуха |
| Миелоидная саркома (хлорома, гранулоцитарная саркома) |
| Миеломная болезнь |
| Миелофиброз |
| Нарушения коагуляционного гемостаза |
| Наследственная a-fi-липопротеинемия |
| Наследственная копропорфирия |
| Наследственная мегалобластная анемия при синдроме Леш — Найана |
| Наследственные гемолитические анемии, обусловленные нарушением активности ферментов эритроцитов |
| Наследственный дефицит активности лецитин-холестерин-ацилтрансферазы |
| Наследственный дефицит фактора X |
| Наследственный микросфероцитоз |
| Наследственный пиропойкилоцитоз |
| Наследственный стоматоцитоз |
| Наследственный сфероцитоз (болезнь Минковского-Шоффара) |
| Наследственный эллиптоцитоз |
| Наследственный эллиптоцитоз |
| Острая перемежающаяся порфирия |
| Острая постгеморрагическая анемия |
| Острые лимфобластные лейкозы |
| Острый лимфобластный лейкоз |
| Острый лимфобластный лейкоз |
| Острый малопроцентный лейкоз |
| Острый мегакариобластный лейкоз |
| Острый монобластный лейкоз |
| Острый промиелоцитарный лейкоз |
| Острый промиелоцитарный лейкоз |
| Острый эритромиелоз (эритролейкоз, болезнь Ди Гульельмо) |
| Отдельные формы лейкозов |
| Пароксизмалъная холодовая гемоглобинурия |
| Пароксизмальная ночная гемоглобинурия (болезнь Маркьяфавы-Микели) |
| Парциальная красноклеточная аплазия |
| Патологическая анатомия поражения оболочек |
| Плазмоклеточный острый лейкоз |
| Полиорганная недостаточность |
| Поражение нервной системы |
| Порфирии |
| Принципы разделения злокачественных и доброкачественных опухолей системы крови |
| Приобретенные геморрагические коагулопатии |
| Причины гемобластозов |
| Пролимфоцитарный лейкоз |
| Ретикулез (ретикулогистиоцитоз, нелипидный ретикулоэндотелиоз, болезнь Абта-Леттерера-Сиве) |
| Серповидно-клеточная анемия |
| Серповидно-клеточная анемия |
| Синдром Дайемонда — Блекфана |
| Сублейкемический миелоз |
| Т-клеточный лейкоз-лимфома взрослых |
| Талассемия |
| Талассемия |
| Тромбофилий, связанные с дефицитом антитромбина III |
| Тромбоцитопатии |
| Тромбоцитопении |
| Фолиеводефицитная анемия |
| Хроническая лучевая болезнь |
| Хронический лимфолейкоз |
| Хронический лимфолейкоз (хронический лимфоидный лейкоз) |
| Хронический лимфоцитарный лейкоз |
| Хронический мегакариоцитарный лейкоз |
| Хронический миелоидный лейкоз |
| Хронический миелолейкоз |
| Хронический моноцитарный лейкоз |
| Хронический моноцитарный лейкоз |
| Хронический эритромиелоз |
| Цитостатическая болезнь |
| Энтеропатии и кишечный дисбактериоз |
| Эритремия |
| Эритремия (истинная полицитемия, эритроцитоз, болезнь Вакеза) |
| Эритропоэтическая копропорфирия |
| Эритропоэтическая протопорфирия |
| Эритропоэтические уропорфирии |
| Ювенильный миеломоноцитарный лейкоз |
Что провоцирует / Причины Острого промиелоцитарного лейкоза:
Причиной острого промиелоцитарного лейкоза, является хромосомная транслокация t (15;17) ведущая к соединению гена рецептора ретиноевой кислоты (RAR-aльфа) c геном опухолевого супрессора PML , продукт которого образует в ядре специфические матрикс-ассоциированные ядерные тельца PML. Цитогенетический анализ выявляет в клетках больных APL транслокации, захватывающие хромосомы 15 и 17. Это специфические транслокации q(15;17) (q22;q11.2) не были вывлены ни в одном другом типе лейкемии миелоцитов или другом злокачественном заболевании. Транслокация (15;17) прерывает RARальфа ген и часть его сливается с локусом PML хромосомы 15, образуя химерный слитый белок PML-RARa. Ген PML кодирует белок, содержащий «цинковые пальцы», и может являться важным транс- действующим транскрипционным фактором в процессе дифференцировки гранулоцитов.
Предполагается, что химерный белок PML/RAR-a инактивирует по доминантно-негативному механизму апоптогенную функцию нормального белка PML, образуя с ним гетеродимеры. Механизмы индукции апоптоза при гиперэкспрессии PML пока не совсем ясны. Экспрессия химерного белка PML/RAR-a, вызывающая инактивацию нормальной функции белка PML, как и перестройка BCR/ABL, ведет одновременно и к изменениям регуляции клеточного цикла, и к частичному блокированию индукции апоптоза (следует заметить, что в отличие от BCR/ABL перестройка PML/RAR-a вызывает также и блок дифференцировки). В результате многонаправленного характера действия гибридных молекул появляются клетки с повышенным пролиферативным потенциалом и одновременно с устойчивостью к негативным регуляторным сигналам и/или неблагоприятным условиям окружающей среды. Предполагается, что такие изменения могут быть уже достаточными для развития по крайней мере некоторых форм гемобластозов. И, действительно, перестройки BCR/ABL или PML/RAR-a часто являются единственными генетическими изменениями, обнаруживаемыми соответственно при хроническом миелоидном и остром промиелоцитарном лейкозах.
Было идентифицировано множество лейкемия-специфических генов, но в результате слияния генов рецептора ретиноидной кислоты (RAR альфа) и гена лейкемии промиелоцитов (PML) возник новый интересный пример таких генов, приводящих к возникновению острой лейкемии миелоцитов (APL).
Три разных химерных гена PML-RARa, длинный (L), средний (M) и короткий (S) являются результатом различного типа сплайсинга экзонов гена PML при сплайсинге транслоцированного гена RARa. Транс-ретиноидная кислота (ATRA) приводит к выздоровлению больных APL, позволяя предполагать, что в процессе транслокации образуется гормон-связывающий белок. Химерный белок PML-RARa, по- видимому, блокирует дифференцировку миелоидных клеток, а обработка ATRA снимает этот эффект.
Гены, вовлеченные в патологический процесс при APM, по-видимому, приводят к структурным изменениям нормального гена (протоонкогена), и его белковый продукт, действуя на клетку-хозяина, вызывает злокачественное перерождение. Этот белок в норме вовлечен в процессы пролиферации и дифференцировки.
Молекулярные и клинические исследования APL больных выявляют, что клетки больных могут начать дифференцировку под воздействием ATRA. Обнаружение транслокации 15;17 дает хороший прогноз. При терапии ATRA перестройка гена RARa существует 2-3 недели, а затем исчезает; после выздоровления восстанавливается нормальная структура гена RARа. Использование ATRA для восстановления созревания клеток и их дифференцировку в гранулоциты приводит к выздоровлению 85-90% пациентов. Это является первым примером лечения рака человека.
В некоторых случаях больных APL, ген RARa может быть вовлечен в другие транслокации и перестройки. Были выявлены два пациента, один с перестройкой 11;17 , а другой с транслокацией 15;17, но без перестройки гена PML. На обоих пациентов терапия ATRA не подействовала. Наблюдения о необходимости сайтов перед геном PML для взаимодействия с ATRA повышает необходимость молекулярной диагностики APL перед назначением или продолжением ATRA терапии. Химерный белок PML-RARa клинически удобен для диагностики и наблюдения при лечении APL.
1.Что такое хронический миелолейкоз?
Хронический миелолейкоз – это заболевание, при котором костный мозг производит слишком много белых кровяных клеток.
Хронический миелолейкоз, или как его еще называют – хронический гранулоцитарный лейкоз – медленно прогрессирующее заболевание крови и костного мозга, которое обычно встречается у людей среднего возраста и редко диагностируется у детей.
В норме костный мозг производит незрелые стволовые клетки крови, которые с течением времени «дозревают». Стволовая клетка крови может стать миелоидной стволовой клеткой или лимфоидной стволовой клеткой. Лимфоидная стволовая клетка в будущем станет белой кровяной клеткой, а миелоидная стволовая клетка – одним из трех типов зрелых клеток крови:
- Эритроцитами, несущими кислород и другие необходимые вещества для всех тканей организма;
- Тромбоцитами, которые образуют сгустки крови, чтобы останавливать кровотечение;
- Зернистыми лейкоцитами — белыми кровяными клетками), которые борются с инфекциями и болезнями.
Таким образом, стволовые клетки крови проходят через несколько этапов развития, чтобы стать эритроцитами, тромбоцитами или лейкоцитами.
При хроническом миелолейкозе слишком большое количество стволовых клеток крови становится одним из видов белых кровяных клеток, которые называются гранулоцитами. Эти гранулоциты – ненормальные клетки, которые не могут стать здоровыми лейкоцитами. Их еще называют лейкозными клетками. Клетки лейкемии могут накапливаться в крови и костном мозге, а значит мешать развитию нормальных лейкоцитов, эритроцитов и тромбоцитов. Когда это происходит, могут начаться анемия, легкое кровотечение или частые инфекции.
Вот так в целом можно объяснить, что же такое хронический миелолейкоз.
Лейкемоидные реакции: диагностика
Перед тем, как приступить к лечению, гематологи ЦЭЛТ проводят диагностику, направленную на выявление этиологического фактора, спровоцировавшего лейкемоидную реакцию. Для этого пациенту назначают:
- Анализы крови, которые, при наличии реакции выявляют повышенный показатель СОЭ и С-реактивного белка, наличие аутоантител. Проводится подсчёт лейкоцитов, определяется соотношение их форм в процентах;
- Посев крови на стерильность, исследование мокрот и урины, направленнее на точное определение возбудителя. Ферментный иммуносорбентный тест выявляет выработанные организмом антитела;
- Рентгенографическое исследования грудной клетки и суставов дают выявляют затемнения и инфильтраты в лёгких при их заболеваниях (в первом случае) и признаки артритов (во втором);
- УЗ-сканирование даёт возможность определить инфекционный мононуклеоз и другие патологии, способные привести к увеличению числа белых кровяных телец;
- Гистология костного мозга выявляет гиперпластические процессы, увеличения количества бластных клеток, а биопсия поражённого лимфоузла – аномальное разрастание коллагена и злокачественную гранулёму.
К каким докторам следует обращаться если у Вас Хронический миелолейкоз:
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Хронического миелолейкоза, ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору – клиника Eurolab всегда к Вашим услугам! Лучшие врачи осмотрят Вас, изучат внешние признаки и помогут определить болезнь по симптомам, проконсультируют Вас и окажут необходимую помощь и поставят диагноз. Вы также можете вызвать врача на дом. Клиника Eurolab открыта для Вас круглосуточно.
Как обратиться в клинику:
Телефон нашей клиники в Киеве: (+38 044) 206-20-00 (многоканальный). Секретарь клиники подберет Вам удобный день и час визита к врачу. Наши координаты и схема проезда указаны здесь. Посмотрите детальнее о всех услугах клиники на ее персональной странице.
Лечение хронического лейкоза
Лечение больных хроническим лейкозом зависит от типа заболевания и прогностических факторов.
Лекарственный метод является основным при лечении хронического лейкоза.
Лечение хронического лимфоцитарного лейкоза (лимфолейкоза) в зависимости от группы риска
Выбор тактики лечения больных ХЛЛ зависит как от распространенности опухолевого процесса, так и от наличия определенных симптомов. С учетом этих факторов, а также клеточных и хромосомных изменений, больные подразделяются на группы риска.
Группа низкого риска.
Прогноз (исход) заболевания у пациентов этой группы благоприятный. Средняя выживаемость составляет 20-25 лет. Обычно лечение не назначается, а рекомендуется тщательное наблюдение. Лишь в случае дальнейшего развития болезни или появления неприятных симптомов применяется лечение.
Группа промежуточного и высокого риска.
У больных при отсутствии симптомов от лечения можно временно воздержаться. При появлении признаков прогрессирования заболевания или новых симптомов может быть назначена терапия.
Химиоптерапия проводится, как правило, противоопухолевым препаратом хлорамбуцилом. При появлении выраженных побочных эффектов этот препарат может быть заменен на циклофосфамид. Иногда используются стероидные препараты (преднизон).
У некоторых больных применяют комбинированную химиотерапию с включением циклофосфамида, доксорубицина, винкристина.
Флюдарабин применяется обычно при рецидиве (возврате) заболевания после лечения комбинацией препаратов. У молодых больных этот препарат можно применять в самом начале лечения. При увеличении селезенки или лимфатических узлов возможно назначение лучевой терапии в низких дозах. В случае появления выраженных симптомов, связанных со значительным увеличением селезенки, выполняется удаление селезенки.
Больным с большим количеством лейкоцитов, нарушающим кровоток, до химиотерапии показан лейкаферез (удаление избытка лейкоцитов, включая опухолевые клетки). Эффект наступает быстро, но бывает временным.
В редких случаях применяется трансплантация стволовых клеток, однако эффективность данного метода еще не доказана.
Иногда ХЛЛ может трансформироваться (превращаться) в острый лейкоз или агрессивную неходжкинскую лимфому (лимфосаркому).
Лечение хронического миелоидного лейкоза (миелолейкоза) в зависимости от фазы заболевания.
Выбор тактики лечения больных с ХМЛ зависит от фазы заболевания (хроническая, акселерации, бластный криз), возраста больного, прогностических факторов и наличия подходящего донора.
Хроническая фаза.
Применение препарата гливек (иматиниб) приводит к достижению полного эффекта у 90% больных ХМЛ.
До этого использовалась химиотерапия высокими дозами препаратов в сочетании с тотальным облучением и трансплантацией стволовых клеток.
Фаза обострения.
Применение гливека может привести к достижению ремиссии (отсутствию признаков болезни), однако период улучшения длится недолго. Использование интерферона также не позволяет получать длительные ремиссии. У 20% больных отмечается положительный ответ на химиотерапию, но он длится не более 6 месяцев.
Приблизительно 15% больных в этой фазе ХМЛ живут в течение нескольких лет после трансплантации стволовых клеток. Эту процедуру лучше выполнять молодым больным после эффективной химиотерапии.
Бластный криз.
В этой фазе болезни опухолевые клетки напоминают таковые при остром миелоидном лейкозе (ОМЛ), причем они мало чувствительны к химиотерапии. Если же и достигается положительный эффект, то он бывает кратковременным. В этом случае возможно применение трансплантации стволовых клеток.
У некоторых больных опухолевые клетки напоминают клетки острого лимфобластного лейкоза (ОЛЛ), которые более чувствительны к химиотерапии. Поэтому использование винкристина, доксорубицина и преднизона может привести к ремиссии.
При поражении центральной нервной системы у больных ХМЛ применяется цитарабин, вводимый в спинномозговой канал, или облучение головного мозга.
Сопоставимые онкогены
Действие вируса лейкемии мышей Абельсона , ретровируса , вызывающего лейкоз на В-лимфоциты мышей, сравнимо с онкогенным эффектом изменения гена в хромосоме Филадельфии . Здесь также ген ABL ( v-ABL ) заменяется другим геном, в данном случае геном вируса GAG , и стимулируется к повышенной активности тирозинкиназы. Из-за сходства генетической модификации соответственно заболевшие мыши используются в качестве модельных организмов для разработки препаратов против онкогенных эффектов хромосомной модификации в фармацевтических исследованиях, а также для фундаментальных исследований.
Фазы хронического миелолейкоза
- Хроническая фаза. В этой фазе находится большинство пациентов, которые обращаются к врачу (около 85%). Средняя продолжительность – 3 – 4 года (зависит от того, насколько своевременно и правильно начато лечение). Это стадия относительной стабильности. Пациента беспокоят минимальные симптомы, на которые он может не обращать внимания. Иногда врачи выявляют хроническую фазу миелолейкоза случайно, при проведении общего анализа крови.
- Фаза акселерации. Во время этой фазы патологический процесс активируется. Количество незрелых белых кровяных телец в крови начинает быстро нарастать. Фаза акселерации является как бы переходной от хронической к последней, третьей.
- Терминальная фаза. Финальная стадия болезни. Возникает при нарастании изменений в хромосомах. Красный костный мозг практически полностью замещается злокачественными клетками. Во время терминальной стадии пациент погибает.
Общее описание
 Хронический миелолейкоз (ХМЛ) — это злокачественное опухолевое заболевание кроветворной системы, поражающее практически все делящиеся клетки миелопоэза, что подтверждается наличием в них аномальной, т.н. филадельфийской (Ph) хромосомы. ХМЛ страдают не более 2-х человек из каждых ста тысяч населения, в основном, это люди зрелого возраста. Лица мужского и женского пола болеют одинаково часто.
Хронический миелолейкоз (ХМЛ) — это злокачественное опухолевое заболевание кроветворной системы, поражающее практически все делящиеся клетки миелопоэза, что подтверждается наличием в них аномальной, т.н. филадельфийской (Ph) хромосомы. ХМЛ страдают не более 2-х человек из каждых ста тысяч населения, в основном, это люди зрелого возраста. Лица мужского и женского пола болеют одинаково часто.
Факторами, способствующими возникновению ХМЛ, считается радиоактивное излучение и генетическая детерминация. К моменту верификации диагноза число Ph-позитивных клеток при цитогенетическом исследовании костного мозга составляет более 95%, т.е. практически все зрелые клетки периферической крови являются ее потомками. При этом гиперплазия затрагивает, в основном, клетки гранулоцитарного ряда, что приводит к лейкоцитозу в периферической крови — от миелобластов до сегментированных нейтрофилов, базофилов и эозинофилов. В дальнейшем происходит миелоидная инфильтрация печени и селезенки. В течении ХМЛ различают хроническую фазу, фазу акселерации и фазу бластного криза. Порядка 85% случаев заболевания диагностируется в его хроническую фазу, в 10% — в фазу акселерации, в 5% — в фазу бластного криза.
Патогенез хронического миелолейкоза
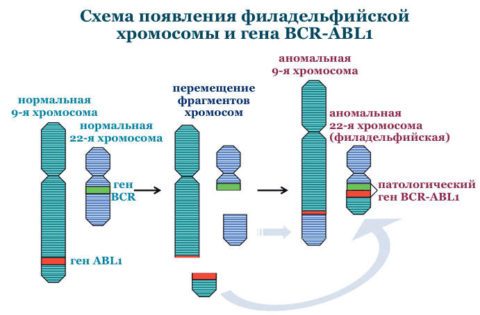
Патогенез хронического миелолейкоза
Гибридный ген BCR-ABL 1, образованный в результате транслокации хромосом, продуцирует синтез белка BCR-ABL. Данный белок представляет собой тирозинкиназу, которая в норме способствует передаче сигнальных импульсов для роста клетки. Созданная путём мутации тирозинкиназа становится активным фактором пролиферации клеток, они начинают делиться и распространяться уже независимо от факторов роста. Происходит процесс создания клонов мутировавшей клетки.
Бесконтрольное деление сопровождается нарушением апоптоза — запрограммированной гибели клеток. Также гибридная тирозинкиназа подавляет естественные функции восстановления в молекулах ДНК, создавая предпосылки для последующих мутаций, что усугубляет патологический процесс.
Размножающиеся клетки являются незрелыми, бластными предшественниками полноценных элементов крови. Постепенно бластные клетки вытесняют функциональные эритроциты, тромбоциты и лейкоциты. Добавляются нарушения и в других хромосомах, что запускает ускоренный процесс разрушения организма в целом.
Номенклатура [ править ]
Филадельфийская хромосома обозначается Ph (или Ph ‘) хромосомой и обозначает укороченную хромосому 22, которая кодирует гибридный ген / протеинкиназу BCR-ABL. Он возникает в результате транслокации, называемой t (9; 22) (q34.1; q11.2) , между хромосомой 9 и хромосомой 22, при этом разрывы происходят в области (3), полосе (4), поддиапазоне ( 1) длинного плеча (q) хромосомы 9 и области (1), полосы (1), поддиапазона (2) длинного плеча (q) хромосомы 22. Следовательно, точки разрыва хромосомы записываются как (9q34. 1) и (22q11.2), соответственно, с использованием стандартов ISCN.
Причины острого миелоидного лейкоза
К факторам риска, способствующих развитию хромосомных нарушений, которые и считаются непосредственной причиной развития ОМЛ, принято относить:
- неблагоприятную наследственность;
- ионизирующее излучение;
- контакт с некоторыми токсическими веществами;
- прием ряда лекарственных препаратов;
- курение;
- болезни крови.
В ходе исследований также было выяснено, что при синдроме Блума, анемии Фанкони, синдроме Дауна вероятность возникновения острого миелоидного лейкоза также увеличивается.
Нельзя не отметить и наследственную предрасположенность при отсутствии генетических заболеваний. Например, если у ближайших родственников был диагностирован ОМЛ, то риск возникновения болезни повышается в 5 раз. Наиболее сильно этому подвержены однояйцевые близнецы — риск составляет 25%.
К каким докторам следует обращаться если у Вас Сублейкемический миелоз:
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Сублейкемического миелоза, ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору – клиника Eurolab всегда к Вашим услугам! Лучшие врачи осмотрят Вас, изучат внешние признаки и помогут определить болезнь по симптомам, проконсультируют Вас и окажут необходимую помощь и поставят диагноз. Вы также можете вызвать врача на дом. Клиника Eurolab открыта для Вас круглосуточно.
Как обратиться в клинику:
Телефон нашей клиники в Киеве: (+38 044) 206-20-00 (многоканальный). Секретарь клиники подберет Вам удобный день и час визита к врачу. Наши координаты и схема проезда указаны здесь. Посмотрите детальнее о всех услугах клиники на ее персональной странице.